MotifLab User ManualThis manual will mainly focus on more in-depth explanations of the different parts of MotifLab. For a more practical introduction on how to use MotifLab, please take a look at the video tutorials. If you have any questions regarding the use of MotifLab that are not answered in this manual or in the tutorials, please do not hesitate to contact us.Note that this user manual is still in preparation (last updated 2024-09-02). Some functionality in MotifLab may not yet be documented here, and some very recent features in MotifLab that are documented here may not be available in the released versions. IntroductionMotifLab is a general workbench for transcription factor binding motif discovery and regulatory sequence analysis. MotifLab allows users to discover motifs and predict binding sites for transcription factors using several published motif discovery programs, and additional data (including for instance information about phylogenetic sequence conservation, DNase hypersensitive sites, epigenetic marks and ChIP-Seq peak regions) can be incorporated into the analysis to corroborate or disprove predictions. The results can be analyzed further to e.g. find motifs that are statistically overrepresented compared to an expected distribution or to discover motifs that are over- or underrepresented in one set of sequences compared to another set.MotifLab allows user to create data objects of different types that can be manipulated and analyzed through the use of operations or examined with interactive tools. Graphical User Interface (GUI)IntroductionNavigationConfiguring the visualizationSessionsCommand-line Interface (CLI)Sometimes a user just wants to run a protocol script to perform an analysis and produce a set of output files but is not interested in looking at the results visually. In such cases, it could be preferable to run MotifLab in CLI-mode with a command-line interface. Running in CLI-mode will be more efficient than running in GUI-mode, since MotifLab does not need to spend time- and memory-resources on data visualization and other amenities (such as e.g. undo/redo functionality). Hence, CLI-mode is the preferred mode when analysing very large datasets.The following command will execute MotifLab from a command-line interface, such as cmd.exe in Windows or a UNIX shell:
The -protocol argument (or "-p" for short) specifies a protocol script to execute. This argument is mandatory unless the -help option is used to list the command line options or the -config option is used to configure MotifLab. If the protocol to be executed analyses sequence regions and no sequences are defined within the protocol itself, the -sequences argument ("-s" for short) must be used to specify a file which contains information about which sequences to analyze. This sequence file can either be in FASTA format, BED format, or Location format. If the sequence file is in FASTA format, the location and genome build for each sequence should preferably be specified in the sequence headers (as explained in the description of the FASTA format), since this information would be required in order to import additional data tracks from preconfigured data sources. Alternatively, a default genome build can be defined with the -genomebuild argument. Also, for FASTA sequence files, a DNA Sequence Dataset named "DNA" will automatically be created based on the information in the FASTA-file and this track will then be available for use in the protocol. If the sequences file is in Location format, a DNA track must be explicitly created in the protocol if this type of data is required, for example with the command All output data objects that are created with the output operation during the execution of the protocol will be saved to files after the execution ends. The filename for each such object will be based on the name of the object itself with a file-suffix which depends on the data format used. For example, an output data object named "BindingSites" which contains output of a Region Dataset in GFF-format will be saved to a file called "BindingSites.gff" (unless the -output option is used to specify a different name). If an output data object contains output in many different formats, the suffix will be set to ".txt". Command line optionsThe following table lists all available command line options. Many of these also have an abbreviated form (which takes the same number of arguments as the unabbreviated form!).If the value for an argument contains spaces it must be enclosed in double quotes. E.g.: -protocol "filename with spaces.txt"
Data injectionThe prompt operation can be used in protocol scripts to allow users to specify values for some types of data objects interactively while the protocol is being executed. This makes it possible to run the same protocol with different values for data objects without having to edit the actual protocol file itself. Whenever a prompt command is encountered during a protocol run, MotifLab will halt and ask the user to enter a value for a named data object. The execution of the protocol will not proceed until a satisfactory value has been provided. Although this behaviour is usually fine, it can be impractical if the user wants to run the protocol several times as a batch job.With data injection the user can specify which values to use for data objects directly on the command line before starting MotifLab rather than having to wait for MotifLab to stop and ask. The command line option syntax is:
The dataname should be the name of a data object that is used as the target for a prompt operation in the protocol script (this is required or else the data injection will not take place). If the data object is a Numeric Variable the provided value should be a number, if the data object is a Text Variable the value should be a text string (enclosed in double quotes if it contains spaces). For all other types of data objects, the value should be the name of a file which contains the input for the data object (in default format for the data type). If you want the value of a Text Variable to be read from file, you can use the prefix "file:" in the value. Example:
Whole genome analysisMotifLab was originally designed to perform analyses on a limited set of sequences, such as for instance a set of promoter sequences from co-regulated genes, and it provides efficient data access by keeping all the data in memory at all times. However, this also means that MotifLab might not be capable of handling extremely large datasets (e.g. whole genomes) that do not fit into the amount of memory available. Some researchers would nevertheless like to use MotifLab to process large datasets, for instance to perform genome-wide motif scanning for TF binding sites that could be filtered based on additional information such as conservation and epigenetic modifications. In such cases, it could be necessary to split a chromosome into smaller segments that are analyzed in turn, rather than loading data for the entire chromosome into memory at once. The "Whole genome analysis mode" that can was introduced in MotifLab version 2.0 allows this task to be performed automatically. The user can simply specify a (large) genomic region and MotifLab will split this region into smaller sequence segments and run the protocol in succession on (collections of) these segments until all of them have been processed. The genomic region to analyse is specified with the -sequences argument as usual, but rather than providing the name of a sequence file, the region is defined using the following format:
The first two fields (genome build and region) are required while the rest are optional. However, the order is important, so if you want to specify the overlap, the segment size and collection size must also be included. The genome build should be a genome build identifier known to the system (e.g. "hg18" or "mm9"). The region specifies the chromosome, start coordinate and end coordinate of the region to be analysed in the format "chromosome:start-end". The start coordinate can be omitted and will then default to 1. For example, the sequence argument "hg19,chr20:10000000-20000000" will analyse the region from position 10000000 to 20000000 on human chromosome 20 (from the hg19 build) whereas the sequence argument "hg19,chr20:63025520" will analyse the whole of chromosome 20 (which is 63025520 bp long). The segment size controls the size of the sequence segments that the genomic region should be divided into (defaults to 10Kbp). The collection size decides how many of these sequence segments should be analysed at the same time (for each execution of the protocol script). This defaults to 100 segments. The overlap length defaults to 0 bp but could be necessary to increase in order to avoid problems introduced by the sequence splitting. For example, if a user wants to perform motif scanning on a long sequence region and this sequence is split in two at a location overlapping a potential binding site, this site can no longer be detected (since each of the sequence segments only receives half a site). By setting the overlap length to a value longer than any of the motifs studied, consecutive sequence segments will overlap by this amount and the full binding site will then be present in either one or both of the segments. To accomodate the overlap, the size of each sequence segment will normally be extended by the specified overlap length. This means that each segment starts at a coordinate on the form "start+k*(segment size)". For example, if the user wants to analyse the region "chr20:40000-44999" by splitting it up in segments of length 1000bp with 100bp overlap, the segments will cover the regions "40000-41100", "41000-42100", "42000-43100", "43000-44100" and "44000-44999" (so each segment starts at the same position that it would have started on if the overlap had been 0bp but ends further downstream). However, by declaring the overlap as a negative value, each segment will have the specified size, but the start position is adjusted instead. For example, splitting the same region as before with an overlap set to -100 will result in the sequence segments "40000-41000", "40900-41900","41800-42800","42700-43700","43600-44600" and "44500-44999". Any output objects that are produced during whole genome analysis will be saved to files as usual, but each sequence group that is analysed in turn will result in a separate output file. The names of the output files will be based on the name of the data object as before, but the files will be distinguished by an additional sequence group number before the file-suffix. For example, if a genomic region is split into 387 segments and MotifLab is told to analyse up to 100 of these segments at a time (collection size=100), the files produced for an output object named "BindingSites" (in GFF format) would be "BindingSites_1.gff", "BindingSites_2.gff", "BindingSites_3.gff" and "BindingSites_4.gff" (where the first three files contain results for 100 segments each and the last file contains the results for the remaining 87 segments). The user can then optionally combine these files together using other command-line tools such as e.g. "cat" in UNIX. Note that if the overlap option is in use, the output files could contain overlapping information which would have to be filtered out to remove duplicates if all the files are concatenated. Data TypesThe figure below illustrates the various data types used by MotifLab. The three data types on the left — Sequence, Motif and Module — are sometimes collectively referred to as the basic types, because they represent the fundamental components that most other data types relate to.The Motif data type models the binding sequence preferences of a transcription factor, and the cis-regulatory Module (CRM) type is a higher-order model of a set of transcription factors that bind cooperatively. The Sequence data type contains information about the origin of a sequence segment (such as a gene) and its location within the genome, but it does not contain the actual DNA sequence. This information is rather represented by a DNA Sequence Dataset, which is a subtype of the more general Feature Dataset type that contains information to annotate sequences. The two other Feature Dataset subtypes are Numeric Dataset, which holds a numeric value for each base within a sequence segment, and Region Datasets, which contains a list of regions representing sequence segments with specific properties, such as e.g. genes, repeat regions or transcription factor binding sites. Objects of the three basic data types can be grouped into (homogeneous) Collections which is useful for referring to sets and subsets of objects, they can be clustered into Partitions or they can be associated with numeric or textual data using Maps. MotifLab has a few more specialized data types used to represent DNA Background models, gene Expression Profiles and "Priors Generators", and some simpler data types to hold atomic Numeric and Text variables. Output Data objects hold text documents in various data formats produced by the output operation, and they can also contain additional embedded files, including images. Finally, results produced by different analyses are stored in Analysis objects, with each type of analysis having its own subtype. 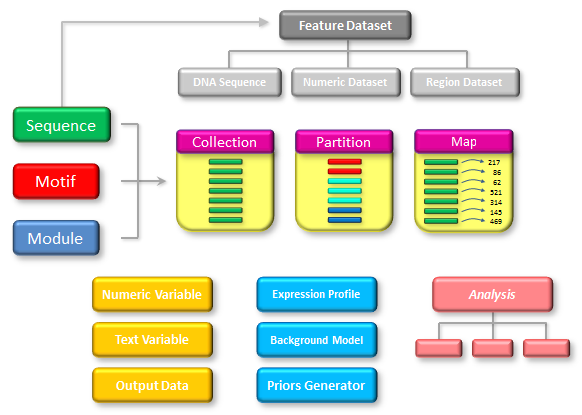
Data objects, names and temporary data objectsEach data object in MotifLab must have a unique name which allows it to be unambiguously identified. Traditionally, the naming conventions for data objects follow the conventions for naming variables in most programming languages, i.e. the name must start with a letter and contain only letters, numbers and underscores. In MotifLab v2 the naming rules for sequences were relaxed a bit to allow sequences to retain names from gene identifiers. This included allowing sequence names starting with numbers (and containing only numbers), and also names containing hyphens, plus-signs, dots, parentheses and brackets.Unlike variable names in most programming languages, however, the data objects in MotifLab can only be referenced through their primary identifier name (or indirectly as part of collections). Hence, data names in MotifLab do not really function like regular variable names, since it is not possible to have two different names referencing the same data object. E.g. if "MA0135" is the name of a motif data object, it is not possible to say "X = MA0135" and then use the name "X" to refer to the the motif "MA0135" later on. If the names of data objects start with underscores, e.g. "_TextVariable1", they are considered as temporary data objects and are given special treatment by MotifLab. Temporary data objects will not be displayed in the GUI in any way, neither in the visualization panel (for sequences and feature datasets) or the data panels (for all data types). When temporary data objects are used in protocol scripts, they will be deleted immediately after the execution of the protocol ends. Temporary data objects can be used for intermediate processing steps whose results are not required to persists beyond the end of the protocol. Sequence
A sequence in MotifLab represents a segment of a DNA strand spanning a specified number of bases.
Usually, a sequence object will represent a "real sequence" where the location of the sequence segment and the genome build it originates from is known.
For example, a sequence could span the segment from position 157,342,949 to position 157,343,321 on the reverse strand of chromosome 2 from the human genome build "hg19".
Alternatively, a sequence object could represent an "artificial sequence" which is not tied to a specific location or genome build (or a "real sequence" whose actual location
or genome build is simply not known). In either case, a sequence object in MotifLab is merely an "empty" template that contains very little information in itself.
Specifically, even though it is referred to as a "sequence", it does not contain information about the actual DNA sequence found at the associated location.
This information is contained in DNA Sequence Datasets, a type of Feature Datasets that can annotate sequence segments with additional information.
The required attributes of a Sequence object is:
A sequence can optionally be associated with a single gene and can then be annotated with the gene's name and the position of the transcription start site and end site.
Creating SequencesSequences are normally created in MotifLab via the "Add Sequences" dialog which can be opened by selecting "Add Sequences" from the "Data" menu or by pressing the double-helix button in the tool bar. In protocol scripts, it is possible to create single (artificial) sequences with specified lengths or (real) sequences defined in BED or Location formats. Multiple sequences can be created with a single command by importing sequence definitions from a Location-, BED- or FASTA-file into the default sequence collection called "AllSequences". The Location-format supports all kinds of sequence metadata (including genome build and location of TSS/TES), but BED-files only contain information about the chromosomal location for each sequence and not its genome build. It is possible to update the genome build for each sequence afterwards, however, with the set[property] command. When importing sequences from FASTA files, the sequence metadata will be included if this information is present in the header of each sequence. If no metadata is present, MotifLab will just create artificial sequences based on the lengths of the sequences found in the FASTA file.Note that even though the FASTA file contains the actual DNA sequences, only the metadata/length of the sequences will be used to create sequence objects. To include the actual DNA sequence you must also create an additional DNA Sequence Dataset based on the same FASTA file. One final way to create new sequences is to extract subsegments from existing sequences with the split_sequences operation.
# Create an "artificial sequence" with length 2000bp and location "chr?:1-2000" from an unknown genome
Seq1 = new Sequence(2000) # Create a new sequence specified in (comma-separated) BED-format with location "chr2:1001-2000" # BED-format arguments: chr,start,end [,gene name,score,strand] Seq2 = new Sequence(chr2,1000,2000) # Create a new sequence specified in (comma-separated) BED-format with location "chr2:1001-2000", # gene name "BRAC1" and reverse orientation. The score attribute in the fifth BED-column is ignored Seq3 = new Sequence(chr2,1000,2000,BRAC1,100,-) # Create a new sequence with location "chr22:36783864-36786063" (reverse strand) from human genome hg19 # associated with the gene "MYH9" with TSS at position 36784063 and TES at position 36677327 # Location-format arguments: Gene name, genome build, chromosome, start, end, TSS, TES, orientation ENSG00000100345 = new Sequence(MYH9, hg19, 22, 36783864, 36786063, 36784063, 36677327, REVERSE) # Same as previous example but the TSS and TES annotations are left out ENSG00000100345 = new Sequence(MYH9, hg19, 22, 36783864, 36786063, - , - , REVERSE) # Create a new sequence spanning 2000bp upstream to 200bp downstream around the transcription start # site of gene "NTNG1" in genome hg18 (gene identifier provided in "HGNC Symbol" format) # Location-format arguments: Gene identifier, identifier type, build, relative start, relative end, anchor Seq4 = new Sequence(NTNG1, HGNC Symbol, hg18, -2000, 200, TSS) # Create a new sequence spanning 100bp upstream to 100bp downstream around the transcription end site # of Entrez gene "56475" from genome build hg18 Seq5 = new Sequence(56475, Entrez gene, hg18, -100, 100, TES) # Create a new sequence spanning the full length of Ensembl gene ENSG00000111249 from hg19 Seq6 = new Sequence(ENSG00000111249, Ensembl Gene, hg19, 0, 0, full gene) # Same as previous example but extended with 500bp additional flanking sequence on both sides Seq7 = new Sequence(ENSG00000111249, Ensembl Gene, hg19, -500, 500, full gene) # Load multiple sequences from file in Location format AllSequences = new Sequence Collection(File:"C:\data\MuscleGenes_-2000+200.txt", format=Location) # Load sequences from file in BED format. The genome build for all the sequences is set afterwards AllSequences = new Sequence Collection(File:"C:\data\genes.bed", format=BED) set AllSequences[genome build] to "mm9" # Create new sequences based on the EnsemblGenes annotations (region track) of the current sequences, # then delete the original sequences. The relationship between new and old sequences is recorded # in the returned SequencePartition SequencePartition1 = split_sequences based on EnsemblGenes. Delete original sequences Modifying SequencesBecause so many other data objects depend on sequences and the locations represented by these objects, sequence objects are usually not allowed to be changed or even renamed after they have been created. Especially, new sequences cannot be created nor can existing sequences be extended after feature datasets have been added (since there would be no feature data for the new sequence segments). However, sequences can still be cropped and dropped.In MotifLab v2, a few sequence properties – namely "genome build", "TSS", "TES", "orientation" and "gene name" – are allowed to be changed after creation. In addition, sequences can be annotated with gene ontology terms and other user-defined properties. The properties of a single sequence can be modified by right-clicking on the name label for a sequence in the Visualization panel (to the left of the tracks visualization) and then selecting "Display sequencename" from the context-menu to bring up a dialog window. Properties for single sequences or collections of sequences can also be updated with the set operation.
# Set the genome build of sequence "Seq1" to "mm9" (this will also update the organism)
set Seq1[genome build] to "mm9" # Set the associated TSS position of sequence "Seq1" to 391829 set Seq1[TSS] to 391829 # Set the "gene name" property of every sequence based on corresponding strings in the Sequence Map NameMap1 set AllSequences[gene name] to NameMap1 # Set the TSS property of every sequence based on corresponding values in the Sequence Numeric Map TSSpos set AllSequences[TSS] to TSSpos # Set the strand orientation of all the sequences in the "Upregulated" collection to the reverse strand set Upregulated[orientation] to "reverse" Using SequencesIndividual sequence objects are rarely used directly in MotifLab, but are rather used as templates for other feature datasets or are referenced to (by name only) as part of collections, partitions and maps. Only a few types of analyses currently make use of information stored directly in sequence objects, such as gene ontology term enrichment analyses.Feature Dataset
Sequence objects are used in MotifLab to refer to specific sequence segments of a genome,
but this data type does not contain any additional information about what is going on at these locations (apart from some metadata).
Further location-specific annotations are kept in feature datasets which come in three different types:
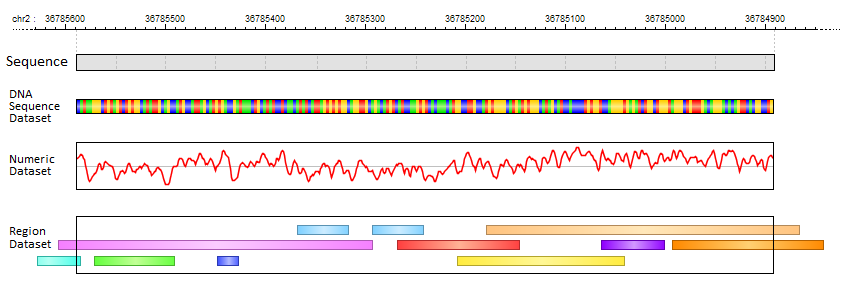 In MotifLab's graphical user interface, all Feature Datasets are listed in the "Features" data panel which is usually located at the top of the left panel, and the feature data tracks
themselves are shown for each sequence in the Visualization Panel. You can configure the visual appearance of feature tracks by right-clicking on a dataset in the "Features" panel
(or selecting multiple by holding down the SHIFT or CONTROL keys) and then select options from the context menu or with keyboard short-cuts.
In MotifLab's graphical user interface, all Feature Datasets are listed in the "Features" data panel which is usually located at the top of the left panel, and the feature data tracks
themselves are shown for each sequence in the Visualization Panel. You can configure the visual appearance of feature tracks by right-clicking on a dataset in the "Features" panel
(or selecting multiple by holding down the SHIFT or CONTROL keys) and then select options from the context menu or with keyboard short-cuts.
DNA Sequence Dataset
DNA Sequence Datasets (also called DNA tracks or DNA sequence tracks) are used to hold the DNA sequence for a sequence segment, represented with one base letter for each position within the sequence.
Most often, objects of this type will hold the original DNA sequence from that location, but this does not have to be the case.
The DNA sequence could instead be a slightly modified version of the original sequence, a scrambled version or even a fully artificially created sequence.
The base letters would normally be either A, C, G or T, but all types of letters are allowed in the sequence. For instance could N's or X's be used to mask portions of a sequence.
Base letters can be in either uppercase or lowercase, and the case may or may not be important depending on the context and the tools used to analyze the sequence.
For example, lowercase letters can be used to indicate repetitive segments of a sequence that should be ignored by a motif discovery tool.
DNA sequences are always stored relative to the direct strand internally in MotifLab (independent of the annotated strand orientation of the sequence), but DNA sequences can be converted on-the-fly to display or manipulate the sequence relative to either strand when necessary. Creating DNA Sequence DatasetsDNA Sequence Datasets are normally imported from predefined tracks or loaded from files (in FASTA or 2bit format), but they can also be artificially created based on a background distribution.
# Import the DNA sequence for the current sequences from the preconfigured track called "Genomic DNA"
DNA = new DNA Sequence Dataset(DataTrack:Genomic DNA) # Import the DNA sequences for the current sequences from a FASTA file. Note that the sequence objects # must already have been created and match the names and lengths of the sequences in the FASTA file. DNA = new DNA Sequence Dataset(File:"C:\data.fas", Format=FASTA) # Create a new 'empty' DNA sequence track consisting of only N's DNA = new DNA Sequence Dataset() # Create a new DNA sequence track consisting of only A's (on the direct strand) DNA = new DNA Sequence Dataset('A') # Create an artificial DNA sequence track by randomly sampling base letters from the distribution # defined in the background model object "EDP_human_3" DNA = new DNA Sequence Dataset(EDP_human_3) Modifying DNA Sequence DatasetsThe main operation for modifying DNA Sequence Datasets is mask, which can replace base letters in certain positions with new letters or change the case of the letters. In addition, the plant operation can insert new binding motifs for transcription factors into an existing DNA sequence.The GUI's draw tool allows users to manipulate the DNA sequence by drawing or typing directly into the visualized track.
# Replace the DNA sequence letters with the letter X within RepeatMasker regions
mask DNA with "X" where inside RepeatMasker # Replace the DNA sequence letters with the letter "A" within RepeatMasker regions # taking the strand orientation of the sequences into account mask DNA on relative strand with "A" where inside RepeatMasker # Change the case of all DNA bases outside of gene regions to lowercase. # Return the result as a new track named "DNA_masked" DNA_masked = mask DNA with lowercase where not inside EnsemblGenes # Replace bases within TFBS regions with new bases randomly sampled from the background model "EDP_human_3" # (This will destroy the binding motifs) mask DNA on relative strand with EDP_human_3 where inside TFBS # Replace bases within TFBS regions with the "sequence" property annotated in these regions mask DNA with TFBS # Insert the motif M00003 at a random location in each sequence (overwriting the current sequence) # Return the modified sequence in a new track called "SequenceWithMotif". # The region track "PlantedMotifs" indicate where the motif was planted in each sequence. [SequenceWithMotif,PlantedMotifs] = plant M00003 in DNA Using DNA Sequence DatasetsDNA sequence tracks are used as input to motif discovery and motif scanning tools (and also module discovery/scanning) and similar operations or tools that search DNA sequences for specific patterns (such as the search and score operations). Background Models can be derived from DNA tracks, and base frequency statistics can also be derived with the statistic operation or the GC-content analysis. Sequence dependent characteristics of the DNA helix, such as e.g. stacking energy and propeller twist, can be derived from a DNA track with the physical operation and represented with numeric tracks. In MotifLab v2 it is possible to extract the corresponding amino acid sequence from the DNA sequence for all six reading frames.DNA sequence tracks can also be referenced in conditions, as demonstrated in the last example below. Here, segments of a DNA sequence masked with X's are used to derive a new Region Dataset representing these masked portions. This is done by first creating a Numeric Dataset with value 1 for every position with an X and then converting this numeric track to a region track.
# Search for the pattern "CACGTG" within the DNA sequence and return matching regions in a new track
Matches = search DNA for "CACGTG" on both strands # Use the MATCH algorithm to scan for matches to JASPAR motifs in the DNA sequence TFBS = motifScanning in DNA with MATCH {Motif collection=JASPAR,Matrix threshold=0.9} # Use the DNA track (on the relative strand) to derive a second-order Markov model of the base distribution BGmodel = new BackGround Model {Track:DNA, Order=2, Strand=Relative} # Count the number of T's in each sequence. Return the result as a Sequence Numeric Map T_count = statistic "T-count" in DNA on relative strand # Derive the GC-frequency from annotated CpG island regions of each sequence GC_content = statistic "GC-content" in DNA where inside CpG_islands # Perform GC-content analysis. Results are returned as an Analysis object rather than a numeric map GC_content = analyze GC-content {DNA track = DNA} # Derive a measure of 'propeller twist' along the DNA helix twist = physical property "propeller twist" derived from DNA using window of size 10 with anchor at center # Derive the amino acid sequence corresponding to the DNA sequence on the direct strand # using a reading frame offset 2bp from the start of the sequence. The AA sequence is returned # as a region track with consecutive 3bp regions named after the amino acids AA_frame2 = extract "Direct-2" from DNA as Region Dataset # Derive a Region Dataset representing the masked regions of a DNA sequence. MaskedRegions = new Numeric Dataset(0) set MaskedRegions to 1 where DNA equals "X" convert MaskedRegions to region where MaskedRegion > 0 Numeric Dataset
Numeric Datasets (also called numeric tracks) represent information with one numeric value for each position within a sequence segment.
The type of information stored in numeric datasets could be, for instance, (per base) phylogenetic conservation levels,
physical or statistical characteristics of the DNA sequence/double helix (e.g. helix twist and roll, or local GC-content), the distance from each sequence position to some target feature,
per base quality scores (for sequence reads), number of ChIP-seq tag counts per position, and position-specific priors used to guide motif discovery, to list but a few examples.
Creating Numeric DatasetsNumeric annotation tracks (based on e.g. data from UCSC Genome Browser or other databases) can be imported from preconfigured tracks or loaded from files in various formats. Numeric tracks can also be derived from information in other types of tracks. For example, Priors Generators can be trained with machine learning methods to predict the location of certain features based on combined information from several different tracks. The output from a Priors Generator is a numeric track where each position reflects a prior probability (or likelihood) that the position could overlap with the target feature (for example a TF binding site).
# Import the "PhastCons100way" annotation track for the current sequences
Conservation = new Numeric Dataset(DataTrack:PhastCons100way) # Import a conservation track from file in WIG format. Conservation = new Numeric Dataset(File:"C:\phastcons.wig", Format=WIG) # Create a new 'empty' numeric track where each position has a value of zero Empty = new Numeric Dataset # Create a new numeric track where each position is assigned the initial value 42 Answer = new Numeric Dataset(42) # Create a new numeric track where the value at each position is the average of the values # from three other tracks AverageValueTrack = combine_numeric track1,track2,track3 using average # Convert the existing region track "CpG_islands" into a numeric track such that all positions # within the original regions are assigned the value 100 and all other position are assigned a value of 0 convert CpG_islands to numeric with value = 100 # Create a new track by counting the number of TFBS regions that overlap with a 5bp window # centered around every position in the track CountTrack = count number of regions in TFBS overlapping window of size 5 with anchor at center # Create a new track where the value in each position is the distance (in bp) # to the closest annotated EnsemblGenes region DistanceToClosestGene = distance from EnsemblGenes # Create a new track based on a measure of predicted 'propeller twist' along the DNA helix twist = physical property "propeller twist" derived from DNA using window of size 10 with anchor at center # Use the TFBSoracle priors generator to derive a new positional priors track based on # an (implicit) set of feature tracks known to the priors generator object TFBS_prior = predict with TFBSoracle Modifying Numeric DatasetsExisting numeric datasets can be modified with arithmetic operations (increase, decrease, multiply and divide) or assigned explicit values with the set operation. They can also be transformed with various mathematical functions (including square root, logarithm and random number), the values could be normalized to a new range or thresholded to create "binary valued" tracks. All of these operations work on a position-by-position basis, but the apply operation will transform tracks with sliding window functions which allow the new value in each position to be derived from values of several positions in a neighbourhood around each sequence position.In addition, the GUI's draw tool allows users to manipulate numeric datasets by drawing directly into the visualized track.
# Increase the values in the Conservation track by 2 for every position
increase Conservation by 2 # Increase the values in the Conservation track by the values from another track (position by position) increase Conservation by DistanceToClosestGeneTrack # Assign the Conservation track a value of 0 within all repeat regions # Return the results in a new track MaskedConservation = set Conservation to 0 where inside RepeatMasker # Return a new track based on the absolute values of Track1 (negative values converted to positive) Track2 = transform Conservation with absolute # Rescale Track1 so that the values fall within the new range 10 to 100. # (i.e. the smallest value in the track will now be 10 and the largest value will now be 100) normalize Track1 from range [dataset.min,dataset.max] to range [10,100] # Transform the Conservation track so that all values previously above (or equal to) 0.5 will be set to 1 # and those below will be set to 0 threshold Conservation with cutoff=0.5 set values above cutoff to 1 and values below cutoff to 0 # Smooth the Conservation track by applying a 25bp wide "Bartlett" sliding window. # This will assign each position a new value based on a weighted average of the values in its vicinity SmoothConservation = apply Bartlett window of size 25 with anchor at center to Conservation Using Numeric DatasetsMotifLab is an expansion of an earlier program called PriorsEditor whose primary purpose was for creating numeric tracks that could be used as position-specific priors to guide the motif discovery process. In addition, apart from being merely descriptive and informative, numeric tracks can be used in conditions to limit operations to certain positions in the sequence or to regions with certain value distributions within their sites.
# Search for motifs and binding sites with MEME using the "Conservation" track as positional priors
[TFBS,MEMEmotifs] = motifDiscovery in DNA with MEME {Positional priors=Conservation, ... } # Mask positions in the DNA sequence with low conservation mask DNA with "N" where Conservation < 0.2 # Remove predicted TFBS regions with low conservation within the site filter TFBS_predicted where region's average Conservation < 0.2 # Use the statistic operation to find the maximum tag count value across all positions in a track. # The result is returned as a Sequence Numeric Map with maximum values for each individual sequence # and with a default map value reflecting the highest count across all sequences Max_tag_count = statistic "maximum value" in ChIPseq_tag_counts # Discover whether TF binding sites are more conserved than other parts of the genome # by analyzing the distribution of conservation track values inside versus outside TFBS regions Analysis1 = analyze numeric dataset distribution {Numeric dataset = Conservation, Region dataset = TFBS} Region Dataset
Region Datasets (also called region tracks) contain sets of regions which are discrete segments of the sequence with associated properties.
Such regions could represent e.g. genes, exons, coding regions, DNase hypersensitive sites, ChIP-seq peak regions, CpG-islands, repeat regions, SNPs and transcription factor binding sites.
Each region has a location within its parent sequence defined by a start and end position, and by extension also a length (which technically could be 0 but not negative)
and genomic location (if the genomic location of the parent sequence is known).
Other standard properties of regions include a type, a numeric score value and a strand orientation (which can be either "direct", "reverse" or "undetermined" and is relative to the genome not the parent sequence).
Additional user-defined properties can be specified for regions as well, like for example the start and end coordinates for CDS subregions of genes or a "sequence" property for TFBS regions denoting the actual binding sequence at the particular site.
These user-defined properties can either have boolean, numeric or textual values.
Regions in the same track may overlap with each other, and regions are also allowed to extend beyond the boundaries of their parent sequence (and could in theory also be located fully outside the sequence). The consequences of regions extending outside of a sequence may differ depending on the particular operation or analysis applied to region tracks. Motif track A motif track is a special kind of region dataset where the type properties of the regions refer to known motifs. Some operations, like motifDiscovery and motifScanning will always return motif tracks, and the motif track status is normally preserved when tracks are manipulated with other operations as well. When region datasets are imported from files or preconfigured tracks, the regions are checked to see if they could potentially correspond to motif sites by comparing the regions' names and lengths to currently defined motifs. If enough regions match with known motifs, the dataset will automatically be converted to a motif track. Region datasets can also be converted to motif tracks manually by right-clicking on a region dataset in the Features Panel and selecting "Convert to Motif Track" from the context menu, or with the following display setting command: $motifTrack(<trackname>)=true. Motif tracks are listed with names in boldface in the Feature Panel in MotifLab's graphical user interface. Module track A module track is a special kind of region dataset where the type properties of the regions refer to known modules. Some operations, like moduleDiscovery and moduleScanning will always return module tracks, and the module track status is normally preserved when tracks are manipulated with other operations as well. When region datasets are imported from files or preconfigured tracks, the regions are checked to see if they could potentially correspond to module sites by comparing them to currently defined modules. If enough regions match with known modules, the dataset will automatically be converted to a module track. Region datasets can also be converted to module tracks manually by right-clicking on a region dataset in the Features Panel and selecting "Convert to Module Track" from the context menu, or with the following display setting command: $moduleTrack(<trackname>)=true. Module tracks are listed with names in bold italics in the Feature Panel in MotifLab's graphical user interface. Nested track A nested track is a special kind of region dataset where the regions may contain nested child regions. For example, in a gene annotation track the top-level gene regions could contain nested regions corresponding to exons within each gene. The module track type described above is actually a kind of nested track where the nested regions correspond to individual motif sites within the module. The extract operation can be used to create new (un-nested) tracks based on only the top-level regions or the child regions of a nested track. Nested tracks are listed with names in italics in the Feature Panel in MotifLab's graphical user interface. Creating Region DatasetsRegion annotation tracks (based on e.g. data from UCSC Genome Browser or other databases) can be imported from preconfigured tracks or loaded from files in various formats. Operations that search for particular patterns within DNA sequences (including motifDiscovery, motifScanning, moduleDiscovery, moduleScanning and search) will usually return the resulting matches as a region track, and regions can also be derived from numeric tracks with the convert operation. The extract operation can extract child regions from a nested track and also extract the start, end and center positions of regions.
# Import the preconfigured "RepeatMasker" annotation track for the current sequences
Repeats = new Region Dataset(DataTrack:RepeatMasker) # Import a region track from file in BED format. Genes = new Region Dataset(File:"C:\RefSeqGenes.bed", Format=BED) # Create a new 'empty' track with no regions Empty = new Region Dataset # Create a new region track based on all the regions from three other tracks AllRegions = combine_regions track1,track2,track3 # The search operation returns a new region dataset with regions matching the search pattern Matches = search DNA for "CAssTG" on both strands # The motifDiscovery operation will return both a Region Dataset (motif track) # with the discovered binding sites and a collection with the newly discovered motifs [TFBS,Motifs] = motifDiscovery in DNA with MEME { ... } # Create a new region track with regions based on consecutive segments in the sequence # with values above 0.8 in the Conservation track ConservedRegions = convert Conservation to region where Conservation > 0.8 # Extract individual TFBS "child regions" from a module track BindingSites = extract "TFBS" from ModuleTrack as Region Dataset # Create a new track with 1bp long regions corresponding to gene transcription start sites # by extracting the first position from each gene region (relative to its own orientation) TSS = extract "regionStart" from EnsemblGenes as Region Dataset Modifying Region DatasetsOperations targeting region tracks will either modify the properties of existing regions, remove regions from the track (filter and prune) or merge regions together. The start and end positions of regions cannot normally be manipulated directly (with e.g. set or arithmetic operations), but some operations like extend can change the size of regions and thereby also alter their location.Most numerical operations that can be used to modify numeric tracks, numeric maps and numeric variables can also be applied to modify numeric properties of regions. Text properties can be altered with the set and replace operations. If the arithmetic operations (increase, decrease, multiply and divide) are applied to text properties of regions, they will function like set operations treating the properties as (comma-separated) lists of values. The increase and multiply operations will then function like set addition (union) whereas the decrease and divide operations will function like set subtraction. However, if arithmetic operations are applied to boolean region properties they function like the following boolean operators: increase = OR, multiply = AND, decrease = NOR, divide = NAND. There are currently no operations that can add new regions to an existing region track, but the GUI's draw tool allows users to draw new regions directly into the visualized track, to delete existing regions and to modify a region's properties in a popup dialog.
# Remove all predicted TFBS regions that are within gene regions
filter TFBS where region inside EnsemblGenes # Remove overlapping TFBS regions representing the same binding motif (as defined in the partition) # and keep only the top scoring region from each cluster prune TFBS remove "alternatives" from MotifPartition1 keep "top scoring" # Reduce the score of TFBS regions by half if they overlap with repeat regions divide TFBS by 2 where region overlaps RepeatMasker # Set the "conservation" property of every TFBS region to the average value from the Conservation track within each site set TFBS[conservation] to average Conservation # Increase the numeric region property "count" by a value defined in the variable for all regions increase TFBS[count] by NumericVariable1 # This command goes through every RepeatMasker region and looks up its type property in the NameMap map # Then it replaces the type of the region with the corresponding value from the map replace NameMap in RepeatMasker property "type" # Increase the size of all DNaseHS regions by 20 bp in both directions extend DNaseHS by 20 # Extend all promoter regions in the upstream direction until they hit the closest gene extend Promoter upstream until inside EnsemblGenes # Merge overlapping ChIPseq regions of the same type into single regions merge similar ChIPseq # Merge all DNaseHS regions located closer than 10 bp apart from each other # (Replace the original regions with a new region beginning at the start of the first region # and ending at the end of the last region) merge DNaseHS closer than 10 Using Region DatasetsThe primary purpose of MotifLab is to predict transcription factor binding sites and cis-regulatory modules within DNA sequences, and region datasets are used to represent such sites. In addition, apart from being merely descriptive and informative, region tracks can be used in conditions to limit operations to certain portions of the sequence. Several different analyses can be applied to region datasets to examine the coverage of the regions in a single dataset, to compare the overlap between two datasets, or to count the number of occurrences of each type of region in a dataset and compare this to another frequency distribution.
# Search for potential transcription factor binding sites in the DNA sequence
# and output the predicted sites in BED format TFBS = motifScanning in DNA with MATCH { ... } output TFBS in BED format # Use the RepeatMasker dataset in a condition to mask only # segments of the DNA sequence that fall within repeat regions mask DNA with "N" where inside RepeatMasker # Count the number of TFBS regions for each motif type and compare these counts to a background # frequency distribution to determine which motifs are overrepresented in this dataset Analysis1 = analyze count motif occurrences {Motif track=TFBS, Motifs=JASPAR, Background frequencies=ExpectedFreq, Significance threshold=0.05, Bonferroni correction="All motifs"} # Count the number of TFBS regions for each motif type within two sequence subsets # representing respectively upregulated and downregulated genes. # Compare these counts between the two sets and use a binomial test to determine # which motifs are over- or underrepresented in one of the sets compared to the other Analysis2 = analyze compare motif occurrences {Motif track=TFBS, Motifs=JASPAR, Target set=UpregulatedGenes, Control set=DownregulatedGenes, Statistical test="Binomial", Significance threshold=0.05, Bonferroni correction="All motifs"} Motif
The Motif data type models the DNA binding recognition sequence of a particular transcription factor (or group of related factors).
Motif scanning tools can be used to predict potential binding sites for different transcription factors by searching DNA sequences for good matches to their corresponding motif models. MotifLab comes bundled with collections of experimentally determined binding motifs from several databases, including TRANSFAC and JASPAR. Novel motifs can also be predicted from sets of sequences with de novo motif discovery tools, or users can define new motifs directly by manually specifying a binding matrix, consensus sequence or explicit list of binding sequences. Motif propertiesThe motif type is one of the richest data types in MotifLab in terms of the amount of different information it can contain.A list of standard motif properties are described below. Except for "ID" and "matrix", all of these are optional. In addition to these, motifs can also have extra user-defined properties.
† These properties are derived from other properties and can not be altered directly. Matrix modelThe main way the binding motif is modelled in MotifLab is with a position-specific scoring matrix (PSSM), sometimes also called a position count matrix (PCM), position frequency matrix (PFM) or position weight matrix (PWM) depending on its format. This matrix is in the form of an N×4 table where each column represents one of the four DNA bases and each of the rows represent one position in the binding motif.A simple count matrix can be created from a set of binding site sequences (aligned and of equal length) by going through each sequence position in turn, counting the number of times each base letter occurs in that position across all the sites and entering this number into the matrix at the corresponding row and column. For example, a matrix derived from the four 6bp binding sequences "CACGTG,CAGGTG,CACGTG,CACGTT" would look like this:
If a matrix is based on a large number of binding sites, the magnitude of the value for a particular base in row i relative to the other bases should approximate well the transcription factor's relative preference towards that base in that position of the binding recognition sequence. A count matrix can be converted into a frequency matrix by dividing the value of each cell with the total sum of the row so that the combined frequencies of the four bases sum to 1.0 for each position. Such a frequency matrix can be further transformed into a weight matrix by replacing each cell value with the log-ratio log( fi,b / pb ), where fi,b is the frequency of base b in position i and pb is the background probability of observing that base in entire genome. A value of 0 for a base b at position i in a weight matrix thus means that the transcription factor shows no particular preference for that base in that position of the recognition sequence (taking the background distribution into account). A positive value reflects a higher preference for that base relative to the other bases and a negative value reflects a lower preference for that base. When MotifLab imports motifs from a file, the matrix models will be kept in their original formats, but MotifLab also tries to detect what kind of format this is so that the matrix can be dynamically converted into other formats if necessary. The rules for determining the format based on the matrix values are:
Consensus modelA secondary way to represent the binding model of a motif is with a consensus string. This is a string of base symbols, one for each position in the binding motif, denoting either single DNA bases or degenerate bases that represent groups of two or more DNA bases with a single symbol.The notation follows the standard suggested by IUPAC:
If a motif already has a matrix model, the correponding consensus string will be derived from that matrix in accordance with the rules outlined in the section below. If the motif has a consensus string but not a matrix model, a matrix will be constructed based on the consensus string. Deriving an IUPAC consensus string from a matrix
To determine the IUPAC symbol to use for a given position in the binding motif, the following rules are tried in order:
Deriving a matrix from an IUPAC consensus string
A consensus string is converted into a frequency matrix by looking up the base symbol at each position i in the table below and assigning the corresponding values to row i in the matrix.
Creating motifs
Motifs are usually generated by motif discovery methods or loaded from pre-defined collections. However, it is also possible to define new motifs manually.
In the GUI, select "Add New ⇒ Motif" from the "Data" menu or press the plus-button in the Motifs Panel and select "Motif" from the drop-down menu. This will bring up the Motif dialog.
The dialog contains multiple tabs where you can enter values for various motif properties. The only required property is the binding motif itself, which can be specified either as a matrix model or a consensus sequence.
Consensus sequences can be entered in IUPAC notation (see above) or as a list of individual binding sequences (separated by any non-letter character). The matrix model will then be created automatically from the consensus sequence.
Note that if you create new motifs that are not part of collections, you must select to display "Motifs" from the drop-down menu in the Motifs Panel in order to see the motifs listed in the panel.
# Creates a new motif named MyMotif with the specified properties
MyMotif = new Motif( Property1:value1; Property2:value2; ... ; PropertyN:valueN ) The only required property is the binding motif itself which can be specified either as an IUPAC consensus sequence ("CONSENSUS" property) or as a matrix model (by setting the properties "A", "C", "G" and "T"). Other standard properties include: SHORTNAME, LONGNAME, CLASS, ORGANISMS, PART, ALTERNATIVES, PARTNERS, QUALITY, FACTORS, EXPRESSION, DESCRIPTION and GO-TERMS (these names must be uppercase). All other specified properties are regarded as being non-standard, user-defined properties. Examples
# Creates the new motif M00001 with IUPAC consensus sequence "srACAGGTGkyG" and short-name "myoD"
M00001 = new Motif(CONSENSUS:srACAGGTGkyG ; SHORTNAME:myoD) # Creates a new motif with a specific matrix model (CACGsG) # The matrix values can be frequencies (like here) or counts M00002 = new Motif( A:0.0,1.0,0.0,0.0,0.0,0.0; C:1.0,0.0,1.0,0.0,0.5,0.0; G:0.0,0.0,0.0,1.0,0.5,1.0; T:0.0,0.0,0.0,0.0,0.0,0.0) Motif manipulationMotifLab v2 introduced several functions to derive new motifs based on existing motifs using the extract operation, including functions to reverse complement a motif, trim bases off the ends or even extend the motif with additional bases. These functions can be applied to both single motifs and collections (the syntax is almost identical in the two cases, except that the names of the single motif functions usually contain the word "motif" somewhere). When transforming a single motif in this way, the result must always be assigned to a new explicitly named motif object. However, when the operation is applied to a collection, the original motifs will be replaced with the new transformed motifs unless you specify a "name suffix" that can be used to derive sensible names for all the new motifs.
# Creates the new motif M00001_RC as the reverse complement of M00001
M00001_RC = extract "reverse motif" from M00001 as Motif # Reverse complements all motifs in the TRANSFAC_Public motif collection (replacing the originals) New_Motifs = extract "reverse" from TRANSFAC_Public as Motif Collection # Takes all the motifs from the TRANSFAC_Public motif collection and creates a reverse complement # motif for each one. The new motifs have names based on the original motifs with the added suffix "_RC" # (e.g. the complement of motif M00001 will be called M00001_RC). The original motifs are kept intact. New_Motifs = extract "reverse; name_suffix=_RC" from TRANSFAC_Public as Motif Collection The following examples demonstrate all of the motif manipulation functions as applied to a full collection. Remember to add the ";name_suffix=X" option after the extract function if you want to create new motifs rather than transforming the current.
# Reverse complements the motifs
New_Motifs = extract "reverse" from TRANSFAC_Public as Motif Collection # Inverts the motifs by reversing the order of the positions (reversing without complementing) New_Motifs = extract "inverse" from TRANSFAC_Public as Motif Collection # Randomly reorders all the rows in each binding matrix New_Motifs = extract "shuffle" from TRANSFAC_Public as Motif Collection # Rounds all the values in the matrix to the nearest integer value # This can be useful for cleaning up count matrices with rounding errors New_Motifs = extract "round" from TRANSFAC_Public as Motif Collection # Expands each motif by adding the bases 'AAA' to the beginning and 'TTT' to the end. # Use a star (*) or the number 0 to denote an empty string if you only want to add to one side. # The resulting motifs will have matrices in frequency format. New_Motifs = extract "flank:AAA,TTT" from TRANSFAC_Public as Motif Collection # Trims each motif by removing 3 bases from the beginning and 4 from the end New_Motifs = extract "trim:3,4" from TRANSFAC_Public as Motif Collection # Trims degenerate flanks from motifs having a specific core surrounded by bases with high variation # It works inwards from both ends and removes bases until it encounters a base # with an IC-content that is greater than or equal to the specified threshold (here 0.5) New_Motifs = extract "trim flanks:0.5" from TRANSFAC_Public as Motif Collection Motif tracksA motif track is a special type of region track where the regions correspond to motif sites. In these tracks the type property of each region site corresponds with the name of a motif. Motif tracks include meta-data properties that specifically tag them as such, and they can be recognized in the Features Panel by having names displayed in boldface font. Also, if you point the mouse at a motif track in this panel, the appearing tooltip will describe the dataset as being a "[Region Dataset, Motif track]".Some operations, like motifDiscovery and motifScanning will always return motif tracks, and if you import a region track from any source, MotifLab will first check if it could potentially be a motif track and mark it as such if at least half of the first ten regions correspond to known motifs. You can also try to manually convert a regular region track into a motif track by right-clicking on a track in the Features Panel and selecting "Convert to Motif Track" from the context-menu. A motif region or motif site is a region within a motif track that represents the location of a transcription factor binding site by having a type property that corresponds to the name of a known Motif model. 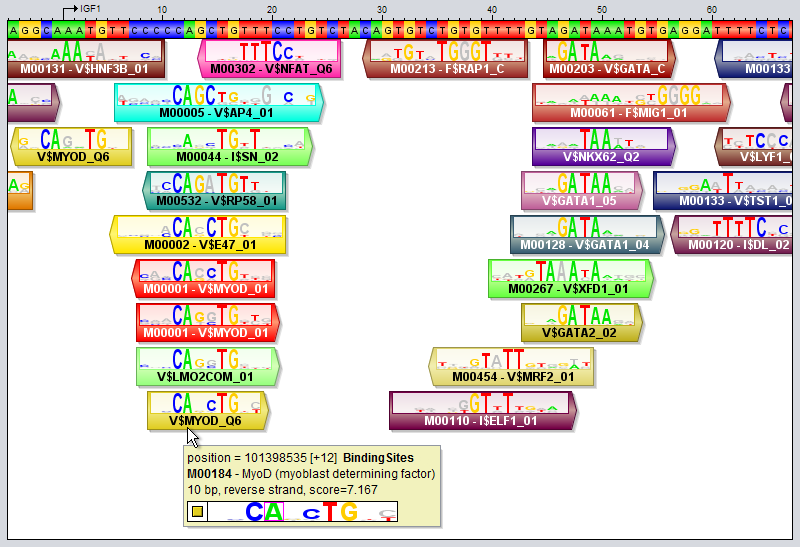
Motif tracks are given special treatment by the GUI's track visualizer, both with respect to how the motif regions themselves are drawn and also how their tooltips are rendered when you point the mouse at a motif region. Motif match logos When the track height and zoom level of the sequence in the sequence window allows it, motif regions will be drawn with motif match logos overlayed on top of the regions. These logos illustrate both the model of the motif itself and how well the model matches the DNA sequence at this particular location. They are inspired by the "Sequence logo" concept introduced by Schneider and Stephens ("Sequence logos: a new way to display consensus sequences", Nucleic Acids Research, Oct 1990, 18(20):6097-6100). The logo is created from the matrix model representation of the motif. For each motif position, the letters for the four bases are first drawn on top of each other. They will be sorted according to their frequency in the model, with the most frequent base on top. Each base letter is also scaled according to its frequency, so if e.g. the frequency of base "G" is 0.46, then the height of the letter G will take up 46% of the stack height, originally. Next, the whole stack of four base letters is scaled once again according to information content, so positions with higher information content (i.e. those whose frequency distribution is more skewed towards a single nucleotide) will have a higher stack. On the other hand, positions that have an almost uniform distribution between the four bases will have a very short height. In addition, the matching base at each position will be colored according to its base's preferred color, whereas the other bases will be drawn in a gray color. Consider as an example the motif region for "M00184 - MyoD" that the mouse cursor is pointing at in the image above. In the last position of this site, the motif model has an almost equal preference for the bases C and T, with C slightly preferred over T (since the most frequent base according to the model - here C - is drawn on top). However, the DNA sequence contains a "T" in this position rather than the most preferred base "C", as indicated by the fact that the T is colored red in the logo while the C is gray. The "colorfulness" of the match logo thus gives an indication of how well a motif model actually matches the DNA sequence at that location. The more tall letters drawn in vibrant colors the logo contains, the better the match between the motif and the sequence. Logos with lots of gray, on the other hand, indicate worse matches. It should be noted that the motif logo colors are not based on the match between the motif logo and the DNA track here seen above the motif track. The DNA sequence used when comparing the motif model to the sequence is taken from a property of the region itself, named "sequence" (this property can be inspected by double-clicking on a region). This "sequence" property is usually set automatically in each region when motif tracks are created based on DNA tracks (using motif discovery or motif scanning tools). Regions that lack this "sequence" property will not be drawn with overlayed motif match logos at all. The visualization of motif sites and their tooltips will differ somewhat depending on whether the motif track is visualized in contracted mode or expanded mode, and the differences between these two modes are described below. You can switch between these modes by selecting a region track in the Features Panel and pressing the X or E keys, or by right-clicking on a track and selecting the mode from the context menu. Expanded Mode In expanded mode (shown in the image above), overlapping motif sites will be drawn beneath each other so that every region is clearly separated from the other regions and distinctly visible in the track.
Contracted Mode In contracted mode, all the regions are visualized on the same line and overlapping regions will thus be drawn on top of each other.
Module
The Module data type (also called composite motif or cis-regulatory module (CRM)) is used to model clusters of binding motifs
that occur in relative proximity to each other and bind multiple TFs that cooperate in regulating one or more genes.
The definition of a module can be loose (e.g. motifs A, B and C should all occur within a span of N bp)
or very strict (e.g. the motifs A, B and C should occur in order with motif B located between 20 to 23 bp after motif A followed by motif C between 35 to 40 bp after motif B; in addition motif B should occur in reverse orientation relative to A and C).
Modules can either be defined manually, they can be discovered "de novo" from sequence data (either DNA tracks or motif tracks) by module discovery programs, or they can be derived based on interaction partner annotations in motifs. Once a collection of modules has been defined, the moduleScanning operation can be employed to search for instances of these modules in either motif tracks or DNA tracks (depending on the particular module scanning program used). Both the moduleDiscovery and moduleScanning operations will return module tracks, which are a special kind of region track where the type property of the regions correspond to module names. The regions of a module track are nested regions where the top-level regions correspond to the full module segment and the child regions correspond to the component motifs of the module. Module definitionThe definition of a module consists of two parts:
A module represents a group of individual binding motifs which are referred to as the component motifs of the module. For example, in a module consisting of binding motifs for the interacting transcription factors SP1, NF-Y and SRF, the component motifs will of course be SP1, NF-Y and SRF. However, in MotifLab these component motifs do not correspond directly to the motif data type. Rather, component motifs represent an intermediate level of "meta-motifs" that are basically sets of equivalent binding motifs for the same TF. The reason for this is that a single TFs can be associated with multiple motif models (for example, the Heat Shock Factor has 12 different motif models in TRANSFAC Public alone!). So, if factor A is represented by N motifs and factor B has M motifs, one can simply define a single module for factors A and B rather than having to define N×M individual modules covering every possible combination of motifs for these two factors.
Module constraints In addition to the component motifs, the module can also be fitted with optional constraints. These constraints can either be global (applying to the module as a whole) or local (applying to a single component motif or the space between two component motifs).
Module propertiesA list of standard module properties are described below. In addition to these, modules can also have extra user-defined properties.
Creating a module in the GUIYou can create a new module by selecting "Add New ⇒ Module" from the "Data" menu or alternatively pressing the "+" button in the Motifs Panel and selecting "Module" from the drop-down menu. Note that the modules you create will only be displayed in the Motifs Panel if the drop-down box above the panel is set to "Modules". Or if the modules are part of collections or partitions you can also see them by selecting these two options.1) Specifying the component motifs In the Module dialog, press the "Add motif" button to add a new component motif to the module. New motifs will be added to the end (right-hand side) of the module. By default, the module will be ordered, which is indicated with angular connector lines between the component motifs. If you uncheck the "Motifs must appear in order" box, the module will be unordered and the connector lines will not be displayed. It is currently not possible to rearrange the order of the component motifs within a module. You can remove a component motif by selecting it and pressing the "remove button". To select a component motif, simply point at the motif box (or above or below it) so that the box border changes to a red color, and then click. The selected portion of the module will be highlighted with a blue background, and all the settings that apply to this component of the module will be enabled in the dialog (such as name, select motifs, color and orientation). 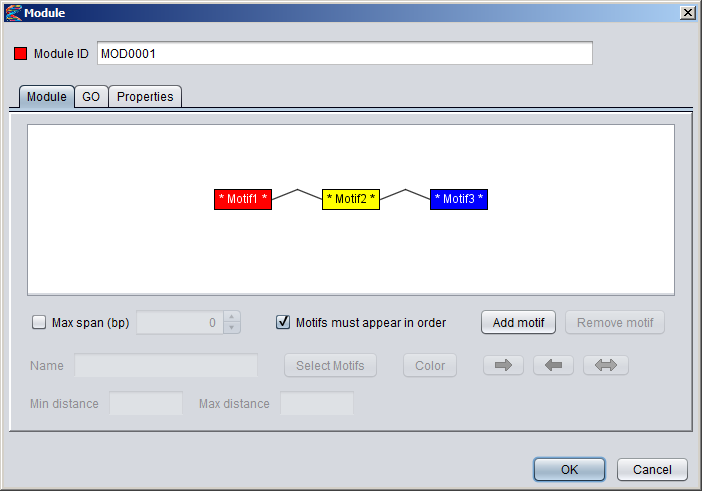
Newly added component motifs will be given generic names on the form "MotifN", and the name will be flanked by stars in the motif box to indicate that the motif has not been associated with any actual motif models yet (e.g. * Motif1 * ). You can change the name of a component motif in the "Name" text field of the dialog and also change the color by clicking the "Color" button. 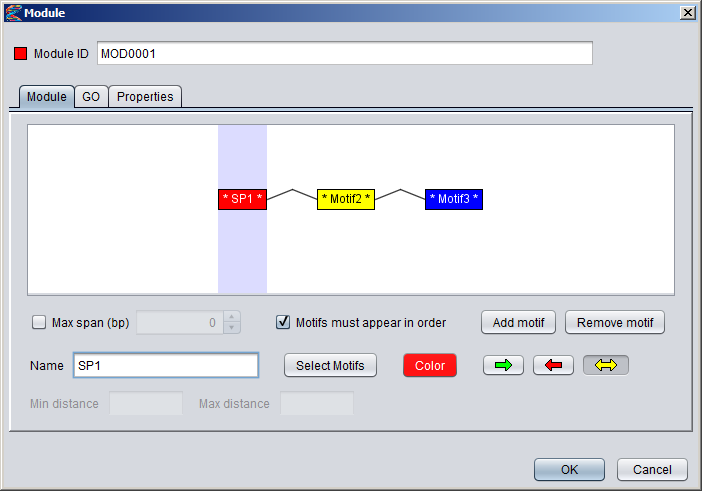
To associate a component motif with actual motif models, either select a component motif and press the "Select motifs" button or double-click on the component motif in the visualization. This will bring up a motif browser where you can select which motifs models to use. Once a component motif has been assigned at least one model, the stars flanking the name in the motif box will disappear. You can hover the mouse over a motif box to see which motif models have been selected for that component. Note that all component motifs must have been assigned at least one basic motif, or else you will not be able to press the "OK" button to close the dialog and create the module. 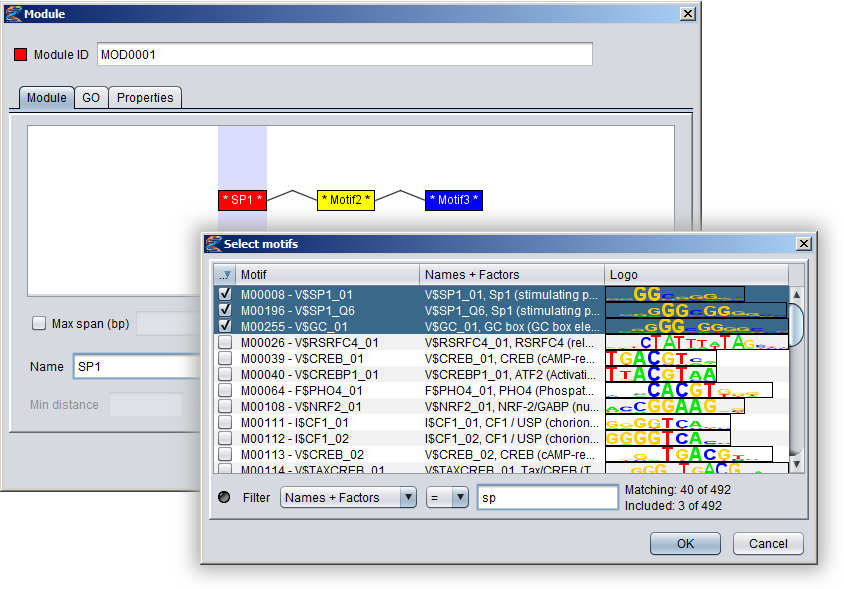
2) Setting distance constraints You can specify a global max length for the module by checking the "Max span (bp)" box and then setting a number in the adjecent field. This constraint is taken to mean that all the component motifs of the module should be located within a sequence window of this size. If the module is ordered you can also specify distance constraints between adjecent pairs of component motifs. To specify such a constraint, simply point to a connector line between motif boxes (the line should turn red), and click to select it. The selected connector should then be highlighted with a blue background, and the settings that apply to this connector will be enabled in the dialog. When you enter numbers into the min distance and max distance fields, these values will appear in brackets above the connector line. It is possible to leave one of the limits blank (either min or max) to say the the distance should be unconstrained in that direction. This will be marked with an asterisk in the brackets, as can be seen for the connector between the NFY and SRF motifs in the figure below. If both limits are left blank, the constraint will be removed. 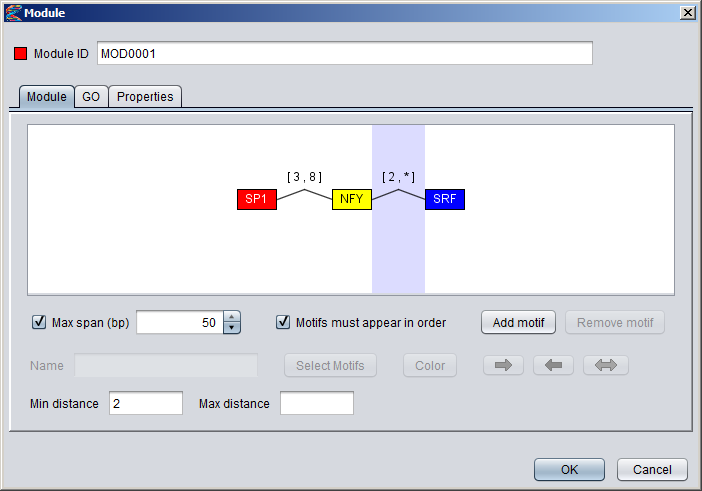
3) Setting orientation constraints It is possible to declare that the component motifs should occur in specific orientations relative to each other. To set an orientation constraint on a component motif, first select it in the visualization and then click on one of the colored arrow buttons underneath the "Add motif" button. If you select the "Direct orientation" button, a green right arrow will also be displayed above the component motif box in the visualization (see motif SP1 in the figure below), and if you select the "Reverse orientation" a red left arrow will be displayed above the motif box (motif SRF in the figure). If you select the yellow "any orientation" bidirectional arrow, the orientation constraint will be removed from the motif and no arrows will be displayed above the motif box (motif NFY in the figure). Note that a direct orientation constraint does not imply that the motif has to be located on the direct strand (and likewise for reverse orientation). It simply means that the underlying motif model must match the DNA sequence in its default (not reverse) orientation, but this could potentially occur on either strand of the DNA sequence. Since orientation constraints are relative, they only make sense if at least two of the component motifs have such constraints. 
Creating a module in a protocolA new module can be created in a protocol script with the following general syntax:
MOD0001 = new Module(... list of property arguments ... )
The arguments are specified as a semicolon-separated list of property definitions, where the name of the property is case-sensitive. The first property argument must be CARDINALITY and its value must match the number of MOTIF arguments. The standard property arguments are described in the table below. Properties that are not in this table are considered to be user-defined properties and must be specified as "propertyname:value" pairs.
Module tracksA module track is a special type of region track where the regions correspond to module sites. In these tracks the type property of each region site corresponds with the name of a module. Module tracks include meta-data properties that specifically tag them as such, and they can be recognized in the Features Panel by having names stylized in both bold and italics. Also, if you point the mouse at a module track in this panel, the appearing tooltip will describe the dataset as being a "[Region Dataset, Module track]".Some operations, like moduleDiscovery and moduleScanning will always return module tracks, and if you import a region track from any source, MotifLab will first check if it could potentially be a module track and mark it as such if at least half of the first ten regions correspond to known modules. You can also try to manually convert a regular region track into a module track by right-clicking on a track in the Features Panel and selecting "Convert to Module Track" from the context-menu. A module region or module site is a region within a module track that represents the location of a cis-regulatory module by having a type property that corresponds to the name of a known Module model. A module region is most often also a nested region where the child regions correspond to the individual TF binding sites that make up the module. These nested regions would then be motif regions whose type properties correspond to names of known Motif models. For example, in the figure below, a module model named MOD0001 is composed of two component motifs – HSF and TATA – with 9 and 6 associated motif models respectively. The particular module site corresponding to this module shown at the top of the track on the right would have the value "MOD0001" for its type-property and two additional properties called "HSF" and "TATA" that would point to two nested motif regions corresponding to the "M00471-V$TBP_01" and "M00147-V$HSF2_01" motif models respectively. (Note, however, that it is technically allowed for a module site to be missing some or all of the component motifs defined in the module). 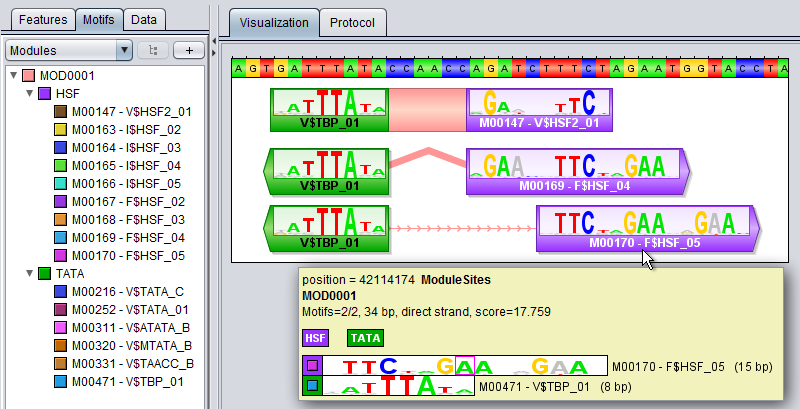
Like motif tracks, module tracks are given special treatment by the GUI's track visualizer, both with respect to how the module regions themselves are drawn and also how their tooltips are rendered when you point the mouse at a module region. In MotifLab version 1.x, the regions of module tracks (and also other nested tracks) would be drawn in two steps. First, a box would be drawn to represent the full module region, and this would be colored according to the chosen color for the module (at least if the "color by type" option was enabled for the track; if not, the module box would be drawn in the selected track color). Second, the individual TFBS of the module (the nested regions) would be drawn on top of this background box in their respective motif colors. An example of this style is shown for the top-most region in the figure above, where the module site spans the full 23bp sequence segment GATTTATAccaaccAGATCTTTCT. The left-hand side of the module site is made up of a TFBS for the TBP factor (green) and the right-hand side is a site for the HSF factor (violet). The middle part "CCAACC" is just inter-motif background sequence where the color of the module itself shines through in pink. The visibility of all module sites corresponding to the same module could be toggled by clicking the colored box in front of the module in the Motifs Panel, and it was also possible to toggle the visibility of the constituent TFBS sites independently of the module by changing the visibility of the motifs. Version 2.0 of MotifLab introduced more ways to visualize modules with different styles of connectors between the component motifs. In addition to the normal background box, modules can now be visualized with straight line segments connecting adjecent motifs, or with angled lines (see second module site in figure above), with curves or with "ribbons". The connector style can be selected by right-clicking on a module track (or other nested track) in the Features Panel and selecting the connector from the context menu. Alternatively, you can select a track (or multiple tracks) in the Features Panel and press the "L" key to cycle through the different connectors. If the "visualize strand (orientation)" option is enabled for a track, the angled line, curved line and ribbon connectors will be drawn pointing upwards if the orientation of the modules correspond with the orientation that the underlying sequence is currently visualized in (i.e. the module is "oriented towards the right-hand side of the screen"). If they have the opposite orientation (module is oriented "towards the left"), the connectors will be drawn pointing downwards. The visualization of module sites and their tooltips will differ somewhat depending on whether the module track is visualized in contracted mode or expanded mode, and the differences between these two modes are described below. You can switch between these modes by selecting a region track in the Features Panel and pressing the X or E keys, or by right-clicking on a track and selecting the mode from the context menu. Expanded Mode In expanded mode, overlapping module sites will be drawn beneath each other so that every region is clearly separated from the other regions and distinctly visible in the track.
Contracted Mode In contracted mode, all the regions are visualized on the same line and overlapping regions will thus be drawn on top of each other.
Collection
Collections are used to refer to (sub)sets of existing data objects or to create/import several new objects with a single operation.
Collections usually always refer to homogeneous sets of data objects of one the three basic data types (motif, module and sequence) and specific subtypes of
collections exist for these types called respectively Motif Collection, Module Collection and Sequence Collection.
Although rarely needed, Text Variables can be used to specify more general collections that are not limited to contain data objects of the basic types.
Creating CollectionsCollections can be created manually by explicitly listing which data objects to include in the collection, or by selecting objects based on some specified criteria. Collections can also be based on or extracted from some other data objects, typically Maps and Analyses. More complex collections can be made by applying set operations (union, intersection etc.) to individual collections. The procedures to create collections described in this section apply to all types of collections. For additional ways to create Motif Collections, Module Collections and Sequence Collections, refer to their respective sections.List of entriesFrom the Collections' GUI dialogs you can select which entries to include by going to the "Manual Selection" tab and checking off the boxes in front of your chosen items (right-clicking on the list will bring up a context-menu with more options to include and exclude items or invert the collection). Alternatively, the "From List" tab lets you to type in the names of items to include and also allows for the use of wildcards and range operators. For example, the star wildcard operator (*) stands for "any string of letters or numbers" so if you enter "MA01*" the collection will include all data items whose names begin with "MA01" (of the relevant type). Many motifs and sequences have names/identifiers on a specific format containing some letters and an incremental number. The colon range operator allows you to specify a subset of items based on a numeric range within the identifier. For example, the range "MA0100b:MA0200b" will include all items whose names start with "MA", ends with "b" and have a number in the middle between 100 and 200 (the prefixes and suffixes around the number are optional but must be the same for all the items, and the numbers need not have the same number of digits). When listing items, the names can refer to either a single basic data object (motif,module,sequence), another collection, or a cluster within a Partition (using the notation "PartitionName->ClusterName"). Note that the "From List" tab allows entries to be separated by either commas, semicolons or spaces/newlines, but in a protocol script they must be separated by commas (they will be converted automatically in "record mode").If you use wildcards, range operators or refer to collections or clusters in the "From List" tab (i.e. refer to multiple data items with one entry), the list can either be parsed and resolved immediately or this can be delayed to when it is first needed ("resolved in protocol"). The second option is now the default behavior but it can be controlled with a checkbox in the "From List" tab. If you choose to "resolve immediately" (by unchecking "resolve in protocol"), then immediately after you press the "OK" button to create the collection, MotifLab will go through all the listed entries to find out exactly which of the currently defined motifs, modules or sequences to include in the collection. This explicit list of basic data objects will then constitute the constructor string for the collection, which is a description of how the collection should be created. This constructor string will be included in the protocol (if you are currently in "record mode") and also as meta-data in the newly created collection itself. (By the way, you can see the constructor for a data object by selecting it in one of the data panels and pressing the "P" key. The constructor will be shown in the log panel.) If you rather choose to "resolve in protocol", the constructor string will instead be the exact text you entered in the "From List" tab (with whitespace and semicolons replaced with commas) prefixed by "List:". The consequence of "resolving immediately" will thus be that entries in the collection are fixed in the protocol even before it is executed, whereas with "resolve in protocol" the final entries in the collection will be decided dynamically when the protocol is run based on the currently defined data objects and contents of other collections.
# Creates a Motif Collection with the motifs M00007, M00013 and M00014
Collection1 = new Motif Collection(M00007, M00013, M00014) # Creates a Motif Collection containing all motifs from the TRANSFAC collection # plus 3 additional JASPAR motifs Collection2 = new Motif Collection(TRANSFAC, MA0004, MA0006, MA0007) # Creates a Motif Collection containing all motifs from the cluster "Upregulated" # within the partition "Significant" Collection3 = new Motif Collection(Significant->Upregulated) # Creates a Motif Collection containing all currently defined motifs Collection4 = new Motif Collection(*) # Creates a Sequence Collection containing all sequences whose names begin with either "ENSG" or "ENSMUS" Collection5 = new Sequence Collection(ENSG*, ENSMUS*) # Creates a Module Collection containing the modules MOD0005, MOD0006, MOD0007 and MOD0008 Collection6 = new Module Collection(MOD0005:MOD0008) Set operationsSet operations can be used to create new collections based on other collections (or single entries or partition clusters). Set operations are processed "left-to-right", so each new entry is processed relative to the collection as it is currently defined by the entries proceeding it. Note that set operators must be placed immediately before the collection it refers to (no space inbetween), and commas must be used between entries in protocols. For example, in a protocol the intersection between collections A and B must be written as "A, &B" and not "A & B".
# Creates a Motif Collection which is the union of motifs in the collections Col1, Col2 and Col3
Collection1 = new Motif Collection(Col1, Col2, Col3) # Creates a Motif Collection containing all motifs present in both Col1 and Col2 (intersection) Collection2 = new Motif Collection(Col1, &Col2) # Creates a Motif Collection containing all motifs present in both Col1, Col2 and Col3 (intersection) Collection3 = new Motif Collection(Col1, &Col2, &Col3) # Creates a Motif Collection containing all motifs from either Col1 or Col2 that are also in Col3 Collection4 = new Motif Collection(Col1, Col2, &Col3) # Creates a Sequence Collection containing all sequences except those in Upregulated Collection5 = new Sequence Collection(*, -Upregulated) # Creates a Sequence Collection containing all sequences except those in Upregulated (using 'complement') Collection6 = new Sequence Collection(!Upregulated) # Creates a Motif Collection containing all motifs that are in either Col1 or Col2 but not in both sets # This XOR operation is accomplished via two intermediate collections (here temporary data objects) _unionSet = new Motif Collection(Col1, Col2) _intersectionSet = new Motif Collection(Col1, &Col2) Collection7 = new Motif Collection(_unionSet, -_intersectionSet) Collections based on propertiesMotifs, modules and sequences have both standard and user-defined properties that can be used to create collections based on a defined condition. You can, for instance, make a collection based on all motifs with IC-content higher than 12.0, or a collection with all sequences that reside on chromosome 2.In the GUI you can create such collections by selecting "Add New ⇒ Collection" from the "Data" menu and then go to the "From Property" tab. The general syntax for creating such collections in protocols is the following:
MyCollection = new <Type> Collection(Property: <property name> <comparator> <target value>)
The property name is selected with an editable drop-down menu in the GUI and it can be enclosed in quotes in the protocol. The available comparator functions vary depending on whether the property is numeric, textual or boolean. The target value can either be a single value or multiple values. In the protocol, multiple values must be separated by commas, and individual values can optionally be enclosed in quotes. In the GUI there is a big text box where you can enter multiple target values which may be separated either by newlines or commas, but values should not be quoted in the GUI.
# Creates a collection containing all motifs with IC-content greater than or equal to 12
Collection1 = new Motif Collection(Property: "IC-content" >= 12) # Creates a collection containing all motifs with sizes in the range 8 to 11 (inclusive) Collection2 = new Motif Collection(Property: "Size" in 8, 11) # Creates a collection containing all motifs belonging to the classes "4.4.1.1" or "2.3.2.0" Collection3 = new Motif Collection(Property: "Classification" equals "4.4.1.1","2.3.2.0") # Creates a collection containing all motifs whose list of associated binding factors # includes names containing the words "CREB" or "NF-Y" as substrings Collection4 = new Motif Collection(Property: "Factors" matches ".*CREB.*",".*NF-Y.*") # Creates a collection containing all motifs whose consensus sequences are listed in the Text Variable Collection5 = new Motif Collection(Property: "Consensus" in TextVariable1) Collections based on values in MapsSimilarly to how collections can be based on data objects having certain values for specific properties, collections can also be based on data objects having certain values in specific maps. At the moment, only numeric maps can be used for this, but support for text maps will be added soon.In the GUI you can create such collections by selecting "Add New ⇒ Collection" from the "Data" menu and then go to the "From Map" tab. The general syntax for creating such collections in protocols is the following:
MyCollection = new <Type> Collection(Map: <map name> <comparator> <target value>)
The name of the map variable is selected with a drop-down menu in the GUI. The available comparator functions are: = , <> , < , <= , > , >= , in The target value should be a single numeric value which can be either a literal string or a numeric data object. If the comparator is "in" the target value should be two numeric values denoting respectively the minimum and maximum value in the range (inclusive). In the protocol the range values must be separated by a comma and enclosed in brackets.
# Creates a collection containing all motifs with a value less than or equal to 0.05 in the "Pvalue" map
Collection1 = new Motif Collection(Map: Pvalue <= 0.05) # Creates a collection containing all motifs with values between 20 and 32 in the "Support" map Collection2 = new Motif Collection(Map: Support in [20, 32]) Random collectionsRandom collections can be constructed with both the new and extract operations, but currently the collection dialogs in the GUI have no way to define them. Hence, you cannot create random collections by selecting "Add New ⇒ Collection" from the "Data" menu, only by extracting random entries from existing collections or by manually typing and executing a new command in the protocol editor. Entries for the new collection can either be sampled from an existing collection or from all currently defined data items of the given type (if no collection is specified). The number of entries to include in the new collection can either be an absolute number or a relative percentage number (value between 0 and 100). If the value is higher than the number of available items, all of them will be included. Non-integer values will be rounded to the nearest integer. Numeric Variables can be used in place of literal numbers.
# Creates a Motif Collection with 10 motifs selected at random from all currently defined motifs
Collection1 = new Motif Collection(Random: 10) # Creates a random Motif Collection containing 10% of the currently defined motifs Collection2 = new Motif Collection(Random: 10%) # Creates a Sequence Collection with 25 random sequences using a Numeric Variable to specify the number NumVar1 = new Numeric Variable(25) Collection3 = new Sequence Collection(Random: NumVar1) # Creates a random Sequence Collection containing one quarter (25%) of the currently defined sequences NumVar2 = new Numeric Variable(25) Collection4 = new Sequence Collection(Random: NumVar2%) # Creates a Motif Collection with 32 entries sampled at random from the JASPAR collection Collection5 = new Motif Collection(Random: 32 from JASPAR) # Creates a Motif Collection with half of the motifs from Collection2 Collection6 = new Motif Collection(Random: 50% from Collection2) # Creates a Sequence Collection by extracting 10 random sequences from the "UpregulatedGenes" collection Collection7 = extract "random 10" from UpregulatedGenes as Sequence Collection # Extracts 10% of the sequences the "UpregulatedGenes" at random Collection8 = extract "random 10%" from UpregulatedGenes as Sequence Collection # Extracts 40 of the sequences in "UpregulatedGenes" using a Numeric Variable to specify the number NumVar3 = new Numeric Variable(40) Collection9 = extract "random NumVar3" from UpregulatedGenes as Sequence Collection # Extracts 40% of the sequences in "UpregulatedGenes" using a Numeric Variable to specify the number NumVar4 = new Numeric Variable(40) Collection10 = extract "random NumVar4%" from UpregulatedGenes as Sequence Collection Importing collections from filesIn the GUI you can import collections from files by selecting "Add New ⇒ Collection" from the "Data" menu and then go to the "Import" tab. Alternatively, you can select "Import Data..." from the "Data" menu and then select your desired collection type from the "Type" drop-down menu in the appearing dialog. The data format of the file is selected from another drop-down menu, and additional format specific argument settings may be defined depending on the format. The file path could either refer to a file on the user's local machine or it could be a URL pointing to a file on the web.The general syntax for creating such collections in protocols is the following:
MyCollection = new <Type> Collection(File: "<file path>", Format=<data format> {<format arguments>} )
The format specific arguments can usually be skipped if default argument values are acceptable. Indeed, the specification of the data format itself can be left out and the default data format for the type will then be assumed (MotifLabMotif for motifs, MotifLabModule for modules and Plain for sequences). There are two ways to view the concept of a collection: A collection can either be thought of as a set of references to other data objects (really just a list of names) or one can view the collection as the set of data objects themselves. The first view is like a shopping list saying e.g. "milk, eggs, oranges" and the second view is like a bag containing the actual groceries. This distinction is important to consider when importing collections from files, because, depending on the data format, the files could contain the actual data objects or just their names. If a file contains descriptions of the basic data objects (motifs,modules,sequences) in sufficient detail, then importing the file will also create these objects in addition to creating the collection object. However, if the file only contains names of motifs, modules or sequences, MotifLab assumes that these data objects must already exist and just creates a new collection listing the names in the file (if the data objects do not exist they will not be added to the new collection).
# Imports a file containing JASPAR motifs from a file in MotifLabMotif data format (default for motifs).
# All the motifs described in the file will be created along with a collection named "JASPAR" that contains # references to all of these motifs JASPAR = new Motif Collection(File: "C:\data\jaspar.mlx" ) # Imports a file containing ScerTF motifs from a file in RawPSSM data format. # All the motifs described in the file will be created along with a collection named "ScerTF" that contains # references to all of these motifs ScerTF = new Motif Collection(File: "C:\data\scertf.pssm", Format=RawPSSM ) # Imports a collection with supposedly important motifs from a file in "Plain" data format. # Since this format does not contain descriptions of the motifs themselves but only list their names, # the referenced motifs must already exist in MotifLab (or they will not be included in the collection). Important = new Motif Collection(File: "C:\data\important_motifs.txt", Format=Plain ) # Imports a file containing modules in MotifLabModule data format (default for modules). # Since this data format includes descriptions of the modules and not just a list of their names, # all the modules described will be created in addition to a collection named "myModules" that contains # references to all of these modules. A MotifLabModule file could possibly also contain descriptions # of the motifs within the modules. If this is the case, these motif objects will be created also. # If the file only contains module descriptions but not motif descriptions, MotifLab assumes that the # motifs must already exist (or else the modules will not work properly). MyModules = new Module Collection(File: "C:\data\my_modules.mod" ) # Imports a collection with supposedly important sequences from a file in "Plain" data format # (This is the default data format for Sequence Collections) # Since this format does not contain descriptions of the sequences themselves but only lists their names, # the referenced sequences must already exist in MotifLab (or they will not be included in the collection). ImportantSequences = new Sequence Collection(File: "C:\data\important_sequences.txt" ) # Imports a collection of sequences from a URL in "BED" data format # Since the BED format contains information about the location of the sequences, these sequences will be # created and added to any sequences already existing. Note, however, that new sequences cannot be added # after Feature Datasets have been defined. The new collection is assigned the name "AllSequences" which # is actually the name of the default sequence collection which cannot really be created explicitly. # The command shown below is the only allowed assignment for "AllSequences", but it will only load # new sequences and add them to the current "AllSequences" not delete to current sequences # (i.e. it will not replace "AllSequences" entirely with the sequences in the file) AllSequences = new Sequence Collection(File: "http://somewebsite.com/sequences.bed", Format=BED ) # Imports a collection of sequences from a URL in "Location" data format # Since this format also contains information about the location of the sequences, these sequences will be # created and added to any sequences already existing. Note, however, that new sequences cannot be added # after Feature Datasets have been defined. The new sequences are added to the sequence collection # "Upregulated" and will also be included in the default sequence collection called "AllSequences" Upregulated = new Sequence Collection(File: "http://somewebsite.com/genes.bed", Format=Location ) Modifying CollectionsCollections can only be created with the operations new and extract, and they cannot really be modified after creation. However, you can achieve the same effect by simply creating a new collection with the same name to replace the older one. If you want to alter a collection relative to its current content you can normally use set operations to accomplish this, e.g.Using CollectionsThe main use of collections is to limit the application of operations and analyses to a subset of motifs, modules or sequences. Collections are also used to import or define multiple basic data objects in a single operation. The compare collections analysis will compare two collections to determine their overlap. In the GUI you can control the visualization settings for collections of motifs, modules or sequences by right-clicking on a collection and selecting you preferences from the context menu or via keyboard short-cuts (show/hide, set colors, etc.).
# Search for occurences of motifs from the "TRANSFAC" collection in DNA sequences
TFBS = motifScanning in DNA with SimpleScanner {Motif Collection=TRANSFAC, ... } # Mask repeat regions in the DNA sequence with N's but only for sequences in the "Upregulated" collection mask DNA with "N" where inside RepeatMasker in collection Upregulated # Remove all TFBS regions associated with motifs in the "Insignificant" collection filter TFBS where region's type is in Insignificant # Delete all sequences in the "Downregulated" collection (as well as the collection itself) drop_sequences Downregulated # Determines the overlap between the two collections "Col1" and "Col2" Analysis1 = analyze compare collections{First=Col1, Second=Col2} # Analyses the GC-content of a DNA track for the sequences in the "Upregulated" collection Analysis2 = analyze GC-content{DNA track=DNA, Groups=Upregulated} Sequence Collection
Sequence Collections are a specific subtype of the general Collection data type that can only contain Sequence objects.
Sequence Collections are mainly used to limit the application of operations and analyses to subsets of sequences, but they can also be used to import sequence definitions from a file.
All Sequence Collections will appear in the "Data Objects" panel in MotifLab's GUI. You can create new Sequence Collections by pressing the "+" button in this panel and then selecting "Sequence Collection" from the appearing menu, or you can go to the "Data" menu in the top menu bar and select "Add New ⇒ Sequence Collection". AllSequences (The default sequence collection)MotifLab has a special sequence collection called "AllSequences" which is regarded as the default sequence collection. This collection is always present and it cannot be created or deleted (however, it will be hidden from the "Data Objects" panel in the GUI when it is empty). Neither can its contents be manipulated directly. The "AllSequences" collection will always contain all the currently defined sequences. Newly created sequences will automatically be added to "AllSequences" and deleted sequences will be removed from "AllSequences". Many operations and analyses require you to specify a sequence collection to apply the operation to, but if none are provided, the "AllSequences" collection will normally be assumed and the operation/analysis will thus be applied to all sequences.Note that similar default collections do not exist for motifs and modules (i.e. there are no "AllMotifs" and "AllModules" collections). Hence, if you want to perform motif scanning with all currently defined motifs you must explicitly create a new Motif Collection containing all motifs and then refer to that collection in the motif scanning tool. Although collections are normally regarded as unordered sets they are actually implemented as ordered lists even though the order is almost never an issue. Actually, the "AllSequences" collection is the only collection where the order is used for something in MotifLab, and it is also the only collection whose order can be manipulated. When outputting sequences they are normally output in the order they have in AllSequences, and this is also the order used to visualize the sequences in the GUI. You can reorder sequences in the GUI (and hence in AllSequences) by pointing at a sequence to give it focus and then use the CONTROL key plus ARROW UP or DOWN to move it, or right-click on a sequence and select "Reorder Sequences" from the context menu. Sorting sequences (either with the sort tool or the sort display setting) will also reorder sequences in the AllSequences collection. Creating Sequence CollectionsAs described in the general section on collections, Sequence Collections can be created by explicitly listing the names of sequences to include, using condititions to select sequences based on property values or values in Numeric Maps, or importing sequence collections from files. In addition, Sequence Collections can also be based on sequence statistics as described below.Collections based on sequence statisticsThe statistic operation can be applied to feature tracks to calculate various statistics, such as counting the number of regions in each sequence, finding the largest value for each sequence in a numeric track or counting the number of A's for each sequence in a DNA track. This operation returns a Sequence Numeric Map with values for each individual sequence. As described in the general section on collections, collections can be created based on their values in Numeric Maps, so you can use the statistic operation to create a map and then create a collection from this map. However, it is also possible to perform this in one step and create a Sequence Collection directly from sequence statistics. (Actually, MotifLab will run the statistic operation in the background to create a map, and then use this to create the collection, but this is done automatically and the map is discarded afterwards).In the GUI you can create such collections by selecting "Add New ⇒ Sequence Collection" from the "Data" menu and then go to the "From Statistic" tab. Press the "Select" button to define the statistic function (this will actually bring up the same dialog that is displayed for the statistic operation) and use the other menus to select the comparator function and target value(s). The general syntax for creating sequence collections based on statistics in protocols is the following:
MyCollection = new Sequence Collection(Statistic: (<statistic function>) <comparator> <target value>)
Examples
# Creates a Sequence collection containing all sequences with more than 20 regions in the TFBS track
# This approach uses the statistic operation to create a map and then uses the map to create the collection TFBS_count_map = statistic "region count" in TFBS Collection1 = new Sequence Collection(Map:TFBS_count_map > 20) # Same as above but this time using the statistic constructor directly in the collection Collection2 = new Sequence Collection(Statistic:("region count" in TFBS) > 20) Motif Collection
Motif Collections are a specific subtype of the general Collection data type that can only contain Motif objects.
Motif Collections are mainly used to limit the application of certain operations and analyses to subsets of motifs, but they can also be used to import motifs from a file or return
results from a motif discovery method.
All Motif Collections will appear in the "Motifs" panel in MotifLab's GUI, provided that you have selected to display "Motif Collections" using the drop-down menu in this panel. You can create new Motif Collections by pressing the "+" button in this panel and then selecting "Motif Collection" from the appearing menu, or you can go to the "Data" menu in the top menu bar and select "Add New ⇒ Motif Collection". Creating Motif CollectionsAs described in the general section on collections, Motif Collections can be created by explicitly listing the names of motifs to include, using condititions to select motifs based on property values or values in Numeric Maps, or importing motif collections from files. In addition, MotifLab comes bundled with several predefined collections of motifs that can be imported, and Motif Collections can also be based on sequence support in motif tracks.Predefined Motif CollectionsMotifLab comes bundled with several predefined collections of motifs from publicly available databases, including e.g. TRANSFAC Public and JASPAR. In the GUI you can create such collections by selecting "Add New ⇒ Motif Collection" from the "Data" menu and then go to the "Predefined" tab. This will display a list of available collections. To import motifs from a collection, simply select the collection in the list and press "OK" (or double-click on a collection in the list). When you select a predefined collection from the list, the resulting collection object will automatically be named after the chosen motif collection, as shown in the text box at the top of the dialog. If you want to specify your own name for the new collection, you can change it in the text box before pressing "OK".Importing a predefined collection of motifs will both create the new collection object and also create motif objects for all motifs defined in the collection. If the collection contains motifs with the same name as already existing motifs, the existing motifs will be replaced without notice. The general syntax for importing predefined motif collections in protocols is:
MyCollection = new Motif Collection(Collection: <name>)
Examples
# Imports motifs from the TRANSFAC Public database
Collection1 = new Motif Collection(Collection:TRANSFAC Public) # Imports motifs from the Jaspar Core database Collection2 = new Motif Collection(Collection:Jaspar Core) You can add your own custom motif collections to the "predefined" list by right-clicking on the collection in the motifs panel and selecting "Save As Predefined" from the context-menu. Enter a name for the collection to use in the list and press "OK". Collections based on motif occurrencesMotif collections can be based on motifs that have a certain sequence support in a motif track. By sequence support for a motif we mean the number of sequences that contain at least one occurrence of that particular motif. For example, one could create a collection with motifs that occur in at least 20 sequences or in 80% of all sequences.In the GUI you can create such collections by selecting "Add New ⇒ Motif Collection" from the "Data" menu and then go to the "From Track" tab. First select the motif track from the drop-drop down menu at the top, then select the comparator function (=,<,<=,>,>=,<>,in) and target value from the bottom menus. The target value can be an absolute number or a relative percentage number (in which case the value should be between 0 and 100), and the value can either be a literal number or a numeric data object (Numeric Variable or Motif Numeric Map). The general syntax for creating such motif collections in protocols is the following:
MyCollection = new Motif Collection(Track: <region track>, support <comparator> <target value>)
Examples
# Creates a motif collection containing all motifs that occur in at least 20 sequences in the TFBS track
Collection1 = new Motif Collection(Track:TFBS, support >= 20) # Creates a collection with all motifs that occur in more than 80% of the sequences in the TFBS track Collection2 = new Motif Collection(Track:TFBS, support > 80%) Module Collection
Module Collections are a specific subtype of the general Collection data type that can only contain Module objects.
Module Collections are mainly used to limit the application of certain operations and analyses to subsets of modules, but they can also be used to import modules from a file or return
results from a module discovery method.
All Module Collections will appear in the "Motifs" panel in MotifLab's GUI, provided that you have selected to display "Module Collections" using the drop-down menu in this panel. You can create new Module Collections by pressing the "+" button in this panel and then selecting "Module Collection" from the appearing menu, or you can go to the "Data" menu in the top menu bar and select "Add New ⇒ Module Collection". Creating Module CollectionsAs described in the general section on collections, Module Collections can be created by explicitly listing the names of modules to include, using condititions to select modules based on property values or values in Numeric Maps, or importing module collections from files. In addition, Module Collections can also be based on sequence support in module tracks or be derived from interaction partner annotations in motif objects.Collections based on module occurrencesModule collections can be based on modules that have a certain sequence support in a module track. By sequence support for a module we mean the number of sequences that contain at least one occurrence of that particular module. For example, one could create a collection with modules that occur in at least 20 sequences or in 80% of all sequences.In the GUI you can create such collections by selecting "Add New ⇒ Module Collection" from the "Data" menu and then go to the "From Track" tab. First select the module track from the drop-drop down menu at the top, then select the comparator function (=,<,<=,>,>=,<>,in) and target value from the bottom menus. The target value can be an absolute number or a relative percentage number (in which case the value should be between 0 and 100), and the value can either be a literal number or a numeric data object (Numeric Variable or Module Numeric Map). The general syntax for creating such module collections in protocols is the following:
MyCollection = new Module Collection(Track: <region track>, support <comparator> <target value>)
Examples
# Creates a module collection containing all modules that occur in at least 20 sequences in the Mod1 track
Collection1 = new Module Collection(Track:Mod1, support >= 20) # Creates a collection with all modules that occur in more than 80% of the sequences in the Mod2 track Collection2 = new Module Collection(Track:Mod2, support > 80%) Collections based on known TF interactionsMotifs can be annotated with lists of known interaction partners (i.e. other motifs) for the associated TF, which give rise to networks of motifs for interacting factors. This information can be used to derive modules based on TFs with known interactions. For a pair of motifs, A and B, it is sufficient that one of the motifs has an annotated interaction with the other for MotifLab to regard the two motifs as interacting (i.e. a one-way directional connection is regarded as being equal to a bidirectional connection). In the GUI you can create such collections by selecting "Add New ⇒ Module Collection" from the "Data" menu and then go to the "From Interactions" tab to specify a set of arguments controlling how the collection should be created. Note that this process will not only create a module collection object, but also create all the underlying modules in that collection. All the created modules will be unordered.1) Defining module motifs The first step in the module creation process is to define the potential module motifs (or meta motifs) that can form the constituent motifs of the modules. This is controlled by the "Group" argument, which can optionally specify a Motif Partition with clusters of motifs that should be considered equivalent to each other (e.g. because they represent the same transcription factor). A module motif (either single motif of cluster) is considered to be interacting with another module motif if at least one of the basic motifs in one of the module motifs are interacting with one of the basic motifs in the other module motif. If the "Group" argument is left blank, each module motif will correspond directly to a single basic motif. However, if the motifs are grouped with a Motif Partition, each potential module motif will correspond to a cluster of motifs. If the "Group" argument is not used, it is possible to use the "Motifs" argument to specify a smaller collection of motifs to consider for this step. If both "Group" and "Motifs" are defined, the "Group" argument will take precendence and the "Motifs" argument will be ignored. 2) Selecting module configurations The "Configurations" argument controls how MotifLab should search the motif interactions network to discover modules. To avoid generating an enormous number of modules from transitive interactions, only cliques in the interaction network will be considered. The cliques can be of size 2 or larger.
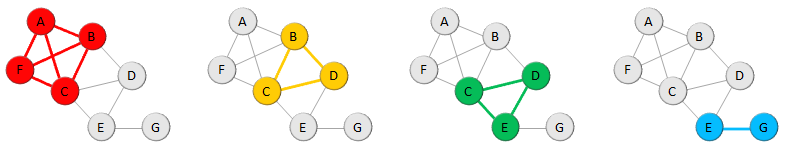
3) Limiting module cardinality If you have selected the "Pairwise" module configuration, all the created modules will have cardinality equal to 2. However, if you have selected the "maximal" or "maximum clique" configurations, the cardinality of the returned modules can be constrained with the "Cardinality limit" option. If this is set to either "at least", "at most" or "exactly", the "Cardinality" argument can specify a number to compare against to constrain the set of returned modules. For example, if "Cardinality limit" is set to "at least" and "Cardinality" to "3", only modules of cardinality 3 or higher will be created. 4) Self-interacting motifs Some transcription factors can interact with other factors of the same type (homo-dimers), so a motif is also allowed to interact with itself. If the "Include self-interactions" option is selected, MotifLab may potentially create modules that consist of pairs of motifs of the same type, e.g. "M1–M1". However, larger modules than that are not allowed to contain duplicate motifs, so if motif "M1" interacts with itself as well as "M2", the modules "M1–M1" and "M1–M2" may be created but not "M1–M1–M1" and "M1–M1–M2". The algorithm that searches for maximal cliques considers cliques that only contain self-interacting motifs to be of size 1, so if M1 interacts with itself as well as M2 and the "maximum clique" configuration is selected, only the "M1–M2" clique will be returned (since this has cardinality 2 whereas "M1–M1" is considered to be of cardinality 1, which is not maximum). However, the "maximal clique" configuration will return both modules. 5) Limiting module span The "Width limit" argument can be used to specify an optional size limit on the module (in bp). If "Width limit" is set to "Total width", the value of the associated "Width" argument defines the maximum length of the module. If "Width limit" is set to "Width per motif", then the width property of each module will be set to the value of the "width" argument multiplied by the cardinality of the module. If "Width limit" is set to "No limit", then no width propery is set. For example, if module M1 consists of 2 component motifs and module M2 of 3 component motifs, then if "Width" is set to 200 (bp) and "Width limit" to "Total width", the width limit of both of these modules is set to 200bp. However, if instead "Width limit" is set to "Width per motif", the width limit of module M1 is set to 200x2=400bp, whereas the width limit of module M2 is set to 200x3=600bp. 6) Limiting collection size If the number of edges in the motif interaction network is large, the number of modules that will be created can potentially be huge. It is possible to limit the size of the returned collection with the "Collection limit" argument. If this is set to a value greater than zero, at most that many modules will be created. The modules are not prioritized in any particular order. In the GUI you can see how many modules MotifLab will create from the motif interactions network with the current argument settings by pressing the "How many modules will be created?" button at the bottom of the dialog. If you consider the number of modules to be too large, you can either set the "Collection limit" to an explicit number or try to limit the number of modules by tweaking the other arguments. The general syntax for creating module collections from interactions in protocols is the following:
MyModules = new Module Collection(Interactions: < ... list of arguments ... >)
Examples
# Creates a module collection with modules based on pairs of interacting motifs.
# Each module will have its global "Max span" constraint set to the cardinality of the module times 50bp Collection1 = new Module Collection(Interactions:Configurations="Pairwise", Width limit="Width per motif",Width=50) # Creates a module collection with modules based on cliques of interacting motifs of size 4 or greater Collection2 = new Module Collection(Interactions:Configurations="Maximal cliques", Cardinality limit="At least",Cardinality=4) Partition
A partition can be thought of as a kind of "super collection" or "collection of collections", or more specifically: a partitioning of data objects into non-overlapping groups called clusters.
Partitions are used to refer simultaneously to multiple non-overlapping subsets/clusters of existing data objects.
Partitions usually always refer to clusterings of data objects of one the three basic data types (motif, module and sequence), and specific subtypes of
partitions exist for these types called respectively Motif Partition, Module Partition and Sequence Partition.
Although rarely needed, Text Variables can be used to specify more general partitions that are not limited to contain data objects of the basic types.
Partitions can, to some extent, be considered as a special case of the Text Map data type, but as Text Maps were only introduced in version 2 of MotifLab, the Partition type predates the more general Text Map.
Creating PartitionsPartitions can be created manually by explicitly listing which data objects to include in which clusters of the Partition, or by clustering data objects automatically based on certain inclusion criteria. Partitions can also be based on or extracted from some other data objects, typically Collections, Maps and Analyses. For additional ways to create Motif Partitions, Module Partitions and Sequence Partitions, refer to their respective sections.Manual clusteringFrom the Partitions' GUI dialogs you can select which entries to include in which clusters by going to the "Manual Entry" tab. Here you can click on entries in the table to select them, and you can hold down the SHIFT and CONTROL keys to select contiguous and non-contiguous ranges respectively. Right-clicking on the table will bring up a context-menu with more options to select items based on different collections. When you have selected a set of entries, right-click on the table to bring up the context menu where you can choose to add the selected entries to an existing cluster or to a new cluster that you will have to name in a popup dialog. Cluster names are case-sensitive and can only contain letters, numbers and underscores (no spaces or special characters). You can also choose to remove the selected entries from their currently assigned clusters. The name of the cluster that each item is assigned to is shown in the second column, and items that have not been associated with a cluster are marked as "unassigned". The "From List" tab lets you to type in comma-separated lists of items to include in each cluster on the format: "<item list> = <cluster name>", and it also allows for the use of wildcards and range operators. For example, the star wildcard operator (*) stands for "any string of letters or numbers" so if you enter "MA01* = <clustername>" the cluster will include all data items whose names begin with "MA01" (of the relevant type). Many motifs and sequences have names/identifiers on a specific format containing some letters and an incremental number. The colon range operator allows you to specify a subset of items based on a numeric range within the identifier. For example, the range "MA0100b:MA0200b" will include all items whose names start with "MA", ends with "b" and have a number in the middle between 100 and 200 (the prefixes and suffixes around the number are optional but must be the same for all the items, and the numbers need not have the same number of digits). When listing items, the names can refer to either a single basic data object (motif,module,sequence), a Collection, or a cluster within another Partition (using the notation "PartitionName->ClusterName"). Note that the "From List" tab allows cluster entries to be separated by either semicolons or newlines, but in a protocol script they must be separated by semicolons (they will be converted automatically in "record mode").It is possible to assign items to the same cluster in separate assignments by reusing the cluster name on the right-hand side of the equals sign. For example, the assignments "MA0005 = Upregulated; MA0006 = Upregulated" will assign both MA0005 and MA0006 to the "Upregulated" cluster. However, each item can only belong to one cluster and it is the last assignment that applies, so the assignments "MA0005 = Upregulated; MA0005 = Downregulated" will assign MA0005 to the "Downregulated" cluster rather than "Upregulated". If you use wildcards, range operators or refer to collections or clusters in the "From List" tab (i.e. refer to multiple data items with one entry), the list can either be parsed and resolved immediately or this can be delayed to when it is first needed ("resolved in protocol"). This can be controlled with a checkbox in the "From List" tab. If you choose to "resolve immediately" (by unchecking "resolve in protocol"), then immediately after you press the "OK" button to create the partition, MotifLab will go through all the listed entries to find out exactly which of the currently defined motifs, modules or sequences to include in each cluster of the partition. The resulting explicit list of basic data objects will then constitute the constructor string for the partition, which is a description of how the partition should be created. This constructor string will be included in the protocol (if you are currently in "record mode") and also as meta-data in the newly created partition itself. (By the way, you can see the constructor for a data object by selecting it in one of the data panels and pressing the "P" key. The constructor will be shown in the log panel.) If you rather choose to "resolve in protocol", the constructor string will instead be the exact text you entered in the "From List" tab (with newlines replaced with semicolons) prefixed by "List:". The consequence of "resolving immediately" will thus be that entries in the partition are fixed in the protocol even before it is executed, whereas with "resolve in protocol" the final clustering in the partition will be decided dynamically when the protocol is run based on the currently defined data objects and contents of other collections and partitions.
# Creates a Motif Partition where the motifs M00007 and M00013 are assigned to a cluster named "First",
# and M00014 and M00015 are assigned to a cluster named "Second". All remaining motifs will be "unassigned" Partition1 = new Motif Partition(M00007, M00013 = First; M00014 = Second; M00015 = Second;) # Creates a Motif Partition where all motifs from the UPREGULATED collection are assigned to the "up" cluster # and all motifs from the DOWNREGULATED collection are assigned to the "down" cluster Partition2 = new Motif Partition(UPREGULATED=up; DOWNREGULATED=down) # Creates a new Motif Partition where all the motifs from the clusters named "Upregulated" and "Downregulated" # within the existing partition named "Significant" are assigned to the "significant" cluster # while all other motifs are assigned to the "nonsignificant" cluster Partition3 = new Motif Partition(*=nonsignificant; Significant->Upregulated,Significant->Downregulated = significant) # Creates a Sequence Partition where all sequences whose names begin with "ENSG" are assigned to the "Human" cluster # and all sequences whose names begin with "ENSMUS" are assigned to the "Mouse" cluster Partition4 = new Sequence Partition(ENSG* = Human, ENSMUS* = Mouse) # Creates a Module Partition where the modules MOD0005, MOD0006, MOD0007 and MOD0008 are assigned to "lower" # and the modules MOD0100, MOD0101, MOD0102 and MOD0103 are assigned to "upper" Partition5 = new Module Partition(MOD0005:MOD0008=lower;MOD0100:MOD0103=upper;) Partitions based on propertiesPartitions can be created by clustering data objects according to the values of certain properties, but this functionality is currently quite limited compared to the same functionality for creating collections. At the moment, only a handful of properties are supported, and clusters can only be made from sequences or motifs having the same value for these properties. Other comparison operators are not supported.For more information, see the corresponding sections under Motif Partitions and Sequence Partitions. Partitions based on values in MapsPartitions can be created by clustering data objects according to their values in Numeric Maps. In the GUI you can create such partitions by selecting "Add New ⇒ <type> Partition" from the "Data" menu and then go to the "From Map" tab.The general syntax for creating such partitions in protocols is the following:
MyPartition = new <Type> Partition(Map: <map name> <comparator> <target value> : <cluster name>)
The "Map:" prefix is followed by one or more cluster assignment rules separated by semicolons. Each assignment rule consists of a map value range defined by a map name, comparator and target value (similar to how collections are created from maps), which is then followed by a colon and a name for the cluster. To define a cluster in the GUI, first select the name of the map variable from the topmost drop-down menu, and then a comparator function from the first drop-down menu behind "Map value". The available comparator functions are: = , <> , < , <= , > , >= , in. Then enter the target value in the third drop-down menu (after the comparator). The target value should be a single numeric value which can be either a literal string or a numeric data object. If the comparator is "in" the target value should be two numeric values denoting respectively the minimum and maximum value in the range (inclusive). In the protocol the range values must be separated by a comma and enclosed in brackets. Finally, enter the cluster name in the text field before pressing the "Add" button. The rule will be added to the large text box at the top of the dialog. To add another cluster, simply enter new values in the drop-down menus and cluster name field and press "Add" again. Note that the rules do not have to refer to the same map. To discard a cluster assignment rule, select it in the large text box and press the "Remove button".
# Creates a partition where motifs having values below 0 in the map Fold are assigned to the cluster "negative",
# motifs with value 0 are assigned to "zero" and those with values above 0 are assigned to "positive" Partition1 = new Motif Partition(Map: Fold<0:negative; Fold=0:zero; Fold>0:positive; ) # Creates a partition where sequences having Rank values below 10 are assigned to "Top", # those with values between 10 and 20 are assigned to "Middle" and those with values above 20 are assigned to "Bottom" Partition2 = new Motif Partition(Map: Rank<10:Top; Rank in [10, 20]:Middle; Rank>20:Bottom; ) Modifying PartitionsPartitions (like collections) can only be created with the operations new and extract, and they cannot really be modified after creation. However, you can achieve the same effect by simply creating a new partition with the same name to replace the older one. In the GUI, you can edit a partition by either double-clicking on it or right-clicking and selecting "Edit ..." from the context menu to bring up the Partition dialog. (As mentioned, this will not actually modify the existing partition, but rather create a new one with the same name.)Using PartitionsPartitions were originally introduced to support clustering operations, although no such operations currently exist in MotifLab. However, external clustering algorithms that return partitions can still be used with the execute operation. The only other operation that returns a Partition is split_sequences, which creates new sequences based on regions in an input region track and returns a SequencePartition where each new sequence created is assigned to a cluster named after the sequence it originated from. Partitions can also be extracted from some other data objects. E.g., the compare motif occurrences analysis groups motifs into clusters based on whether or not they are overrepresented in one of two sequence sets, and these clusters can be extracted as a Motif Partition.Partitions can also be used as arguments to some operations and analyses, typically to apply the operation/analysis to several individual groups in order. For example, the analyses GC-content, Numeric Map distribution, region dataset coverage and benchmark all calculate statistics on various data. By using a partition to group the data into clusters, these statistics will be calculated separately for each cluster. The prune operation can remove overlapping TFBS predictions that represent the same binding motif, and it relies on a Motif Partition to tell it which motifs are considered to be similar.
# Cluster sequences based on sequence similarity using the Starcode program
SequenceGroups = execute Starcode {Sequence Collection=AllSequences, ... } # Extract a Motif Partition with 6 clusters from a Compare Motif Occurrences analysis that compares the frequency of # motif occurrences in two sequence sets. The clusters contain: # 1) the motifs that only occur in the first sequence set # 2) the motifs that occur in both sets but are overrepresented the first set # 3) the motifs that only occur in the second sequence set # 4) the motifs that occur in both sets but are overrepresented in the second set # 5) the motifs that occur with about the same frequency in both sequence sets (overrepresented in neither) # 6) the motifs that do not occur at all in either of the sequence sets Motif_groups = extract "clusters" from Analysis1_UP_vs_DOWN as Motif Partition # Calculate GC-content statistics of a DNA track for each of the clusters in SequencePartition1 GC_analysis = analyze GC-content {DNA track=DNA, Groups=SequencePartition1} # Finds overlapping regions in the TFBS track for motifs that are in the same cluster in the Motif Partition named # 'AlternativePartition' and removes all the duplicates so that only the region with the highest score remains TFBSpruned = prune TFBS remove "alternatives" from AlternativePartition keep "top scoring" Sequence Partition
Sequence Partitions are a specific subtype of the general Partition data type that can only contain Sequence objects.
All Sequence Partitions will appear in the "Data Objects" panel in MotifLab's GUI. You can create new Sequence Partitions by pressing the "+" button in this panel and then selecting "Sequence Partitions" from the appearing menu, or you can go to the "Data" menu in the top menu bar and select "Add New ⇒ Sequence Partition". Creating Sequence PartitionsAs described in the general section on partitions, Sequence Partitions can be created by explicitly listing the names of sequences to include in each cluster, or by using assignment rules to select which sequences to include in each cluster based on their values in Numeric Maps. In addition, Sequence Partitions can also be based on the values of some sequence properties as described below.Partitions based on sequence propertiesSequence Partitions can be created by clustering together sequences that have the same value for a selected sequence property.Currently, only four such sequence properties are supported:
In the GUI you can create such partitions by selecting "Add New ⇒ Sequence Partitions" from the "Data" menu and then go to the "From Property" tab and select the property from the drop-down menu. The general syntax for creating sequence partitions based on properties in protocols is the following:
MyPartition = new Sequence Partition(Property: <property name>)
Examples
# Cluster all the sequences based on genome build
Partition1 = new Sequence Partition(Property: genome build) # Divide the sequences into two groups based on strand orientation Partition2 = new Sequence Partition(Property: strand orientation) Motif Partition
Motif Partitions are a specific subtype of the general Partition data type that can only contain Motif objects.
All Motif Partitions will appear in the "Motifs" panel in MotifLab's GUI, provided that you have selected to display "Motif Partitions" using the drop-down menu in this panel. The Partitions are displayed in a hierarchical fashion with three levels. The top level shows the partitions themselves, the second level shows the clusters and the third level shows the motifs within each cluster. You can create new Motif Partitions by pressing the "+" button in the motifs panel and then selecting "Motif Partitions" from the appearing menu, or you can go to the "Data" menu in the top menu bar and select "Add New ⇒ Motif Partition". Creating Motif PartitionsAs described in the general section on partitions, Motif Partitions can be created by explicitly listing the names of motifs to include in each cluster, or by using assignment rules to select which motifs to include in each cluster based on their values in Numeric Maps. In addition, Motif Partitions can also be based on the values of some motif properties as described below.Partitions based on motif propertiesMotif Partitions can be created by clustering together motifs that have the same value for a selected motif property.Currently, only two such motif properties are supported:
In the GUI you can create such partitions by selecting "Add New ⇒ Motif Partitions" from the "Data" menu and then go to the "From Property" tab and select the property from the drop-down menu. The general syntax for creating motif partitions based on properties in protocols is the following:
MyPartition = new Motif Partition(Property: <property name>)
Examples
# Cluster motifs into equivalence groups based on their alternatives annotations.
# Each cluster will be named after the motif with the shortest name. Partition1 = new Motif Partition(Property: Alternatives) # Cluster motifs into 6 groups based on their top-level TRANSFAC classification Partition2 = new Motif Partition(Property: Class_1_level) # Cluster motifs into groups based on the two topmost levels of the TRANSFAC classification hierarchy Partition2 = new Motif Partition(Property: Class_2_levels) Module Partition
Module Partitions are a specific subtype of the general Partition data type that can only contain Module objects.
All Module Partitions will appear in the "Motifs" panel in MotifLab's GUI, provided that you have selected to display "Module Partitions" using the drop-down menu in this panel. The Partitions are displayed in a hierarchical fashion with three levels. The top level shows the partitions themselves, the second level shows the clusters and the third level shows the modules within each cluster. You can create new Module Partitions by pressing the "+" button in the motifs panel and then selecting "Module Partitions" from the appearing menu, or you can go to the "Data" menu in the top menu bar and select "Add New ⇒ Module Partition". Map
A map is basically a two-column lookup table which associates the name of a data object (or key in the first column) with a corresponding value (in the second column).
There are two general types of maps: Numeric Maps and Text Maps.
Numeric maps associate each data object with a numeric value, whereas values in Text Maps (introduced in MotifLab version 2) can be any text string.
The general Numeric Map type have three specific subtypes – Sequence Numeric Map, Motif Numeric Map and Module Numeric Map
– that hold values for sequences, motifs and modules, respectively.
Likewise, the general Text Map type have the subtypes Sequence Map, Motif Map and Module Map.
Other types of maps can be represented with the Text Variable type.
An example of a Motif Numeric Map that associates motif names with numeric values is shown below.
A data item, such as a motif, that is explicitly included in the map is said to have an assigned value, whereas data items that are not explicitly included in the map are unassigned and will use a default value instead. If you examine a map in the GUI, both assigned and unassigned entries will be shown in the table, but the values of unassigned entries that use the default value will be shown in a gray color rather than a black. Using the map shown above as an example, the MA0005 motif is unassigned, and its value, which defaults to 0, is therefore colored gray in the table. The value for the MA0007 motif is also 0, but this is not the default value since MA0007 is explicitly assigned as indicated by the black color used for the value. If the default value were to be changed to e.g. 3 at a later time, the value of MA0005 would then be 3, but the value of MA0007 would still be 0. Unless otherwise specified, the default value for Numeric Maps will be 0 and the default value for Text Maps will be an empty string. Creating MapsMaps can be created in the GUI by selecting "Add New ⇒ <Type> Map" from the "Data" menu, or by pressing the "+" button in the Data Objects panel menu and selecting the map type from the drop-down menu. All maps of every type will appear in the Data Objects panel.The general syntax for creating maps in protocols is shown below. The values for different data items are specified with a comma-separated list of "key=value" pairs.
MyMap = new <Type> Map(<key1>=<value1>, <key2>=<value2>, ... , <keyN>=<valueN>, _DEFAULT_=<value>)
The key can refer to a collection, in which case all the entries in the collection will be associated with that value. The key can also contain the wildcard symbol (*) that will match any string of characters. In MotifLab v2, keys can refer to clusters within partitions on the form Examples
# Create an "empty" Motif Numeric Map with default value 7.
# All motifs will be unassigned and fall back to the default value. MotifNumericMap1 = new Motif Numeric Map(7) # Create a Motif Numeric Map with default value 0 (implicit) and three motifs with explicitly assigned values. MotifNumericMap2 = new Motif Numeric Map(MA00001=243, MA00002=132, MA00003=193) # Create a Motif Numeric Map where each motif is assigned the value 8. The default value will be 0 (implicit). MotifNumericMap3 = new Motif Numeric Map(*=8) # Create a Sequence Numeric Map where all sequences whose names start with ENSG have value 1 # and those whose names start with ENSMUS have value -1. Other sequences have the default value (0) SequenceNumericMap1 = new Sequence Numeric Map(ENSG*=1, ENSMUS*=-1) # Create a Motif Numeric Map where all motifs in the TRANSFAC collection have value 1 # and all motifs in the JASPAR collection have value 2 MotifNumericMap4 = new Motif Numeric Map(TRANSFAC=1, JASPAR=2) # Create a Module Text Map where all modules in the "C1" cluster in ModPar1 have values "hello world" # and those in the "C2" cluster have values "goodbye world" ModuleMap1 = new Module Map(ModPar1->C1="hello world", ModPar1->C2="goodbye world") # Create a Motif Text Map where all motifs in the range MM0001-MM0199 have the value "first" # and those in the range MM0200-M0299 have the value "second" MotifMap1 = new Module Map(MM0001:MM0199="first", MM0200:MM0299="second") Modifying MapsValues in maps can be set explicitly with the set operation or changed relative to the current values with the arithmetic operations increase, decrease, multiply and divide. Numeric Maps can also be transformed with threshold and transform.When an operation is applied to a whole map, the default value will also be changed accordingly. However, if you only apply the operation to a subset of the entries in the map, the default will not be changed. It is currently not possible to directly change the value of a single named entry in the map. This can only be done by first creating a collection containing only that data item and then limiting the application of a map operation to elements in that collection. Also, the default value of a map cannot be changed directly by itself, but this limitation can also be circumvented with the more cumbersome approach shown in the last example below. Note: When a modifying operation is applied to a map, MotifLab will go through all the applicable entries and recalculate the values before updating each of these entries in the map. This will mean that entries that were previously unassigned may now suddenly become assigned. It you want to avoid this, you will have to limit the operation to only apply to the currently assigned entries as shown in the example below. Likewise, if you output a full map to a file and then import it back again later, entries that were unassigned in the original map will be assigned in the new map. To avoid this you should only output the assigned entries to the file (plus the default value). Examples
# Increase the values of all entries in the map by 3, including the default value.
# Note: Previously unassigned entries in the map will now become assigned. increase MotifNumericMap2 by 3 # Increase the values of all assigned entries in the map by 3. (The default is not changed) # This is done by first creating (extracting) a collection with all currently assigned entries # and then limiting the application of the operation to only the entries in this collection. AssignedEntries = extract "assigned entries" from MotifNumericMap2 as Motif Collection increase MotifNumericMap2 by 3 in collection AssignedEntries # Increase the values of all entries in the map by their corresponding values in a second map increase MotifNumericMap1 by MotifNumericMap2 # Set the default value of the existing map MotifNumericMap2 to 13. # This is accomplished by creating a backup copy of the original map, replacing the original with a new empty map # with the new default value and finally resetting all the entries to their original values from the backup copy. AssignedEntries = extract "assigned entries" from MotifNumericMap2 as Motif Collection backup_copy = copy MotifNumericMap2 MotifNumericMap2 = new Motif Numeric Map(13) set MotifNumericMap2 to backup_copy in collection AssignedEntries Using MapsMaps can be used to store information about e.g. the gene expression levels of you sequences (using a Sequence Numeric Map) or motif match score cutoff thresholds for individual motifs (using a Motif Numeric Map). Some operations, like statistic, will return maps, and the results of many analyses include tables whose individual columns can be extracted as maps. Maps can be used to hold additional information about basic data items (sequences, motifs and modules) similar to user-defined properties for these items. In fact, it is easy to extract a named property from a collection of such data items and return the result in a corresponding map. Likewise, it is just as easy to set (or modify) properties of basic data items to values held in maps.
# General syntax for extracting a property from a collection of basic data items and return the result as a Map
propertyMap = extract "<type>:"<property>" from SomeCollection as <Type of Map> # Even simpler syntax for creating a map of a specified type based on some property propertyMap = new <Type of Map>(Property:propertyname) # Create a Motif Numeric Map containing the IC-content for each TRANSFAC motif motif_IC = extract "motif:IC-content" from TRANSFAC as Motif Numeric Map # Create a Motif Numeric Map with IC-contents for all known motifs motif_IC = new Motif Numeric Map(Property:IC-content) # Create a Sequence Map containing associated gene name for each sequence (if known) geneNames = extract "sequence:gene name" from AllSequences as Sequence Map # Same as above geneNames = new Sequence Map(Property:gene name)
# General syntax to set properties of basic data items based on maps. The two alternatives achieve the same result.
# Note that not all properties are allowed to be altered in this way (e.g. size properties are usually derived) set SomeCollection[propertyname] to SomeMap set SomeCollection property "propertyname" to SomeMap # Add a user-defined numeric property called "score_cutoff" to all TRANSFAC motifs set TRANSFAC[score_cutoff] to MotifNumericMap1 # Increase the "score_cutoff" property of all TRANSFAC motifs by their corresponding value in MotifNumericMap2 increase TRANSFAC property "score_cutoff" by MotifNumericMap2 Using Maps as argumentsWhen you use a map as an argument for operations that handle collections of data objects, the argument will be behave in an intuitive way by taking on the value for the closest naturally associated object in each iteration. For example, if you use a Motif Numeric Map as a "cutoff threshold" argument in a motifScanning operation, the scanning algorithm will use the cutoff value associated with motif X in the map when scanning for hits to motif X, and the value for motif Y from the map will be used as cutoff when scanning for hits to motif Y. On the other hand, if you instead use a Sequence Numeric Map for the same cutoff argument, the value for sequence A in the map will be used as the cutoff when scanning for both motif X and Y in sequence A, but the value for sequence B from the map will be used for all motifs when scanning sequence B. For more information, see here!Numeric Maps
A numeric map is a subtype of the general Map type where the values can only be numeric.
There are three different types of numeric maps – Sequence Numeric Map, Motif Numeric Map and Module Numeric Map
– that hold values for sequences, motifs and modules, respectively.
Numeric maps that contain "Data⇔Value" associations for other types of data can be defined using Text Variables.
Creating Numeric MapsThe general syntax for creating Numeric Maps in a protocol is shown below.
MyNumericMap = new <Type> Numeric Map(<key1>=<value1>, <key2>=<value2>, ..., <keyN>=<valueN>, _DEFAULT_=<value>)
The argument is a comma-separated list of "key=value" pairs where the keys can be the name of a single data object (of the applicable type) or a collection. The wildcard operator (*) is also supported. In MotifLab v2, the key can also be a reference to a partition cluster, or it can refer to a range of data objects. The values on the right-hand side of the assignments can only be numeric constants and not references to other data objects (such as Numeric Variables). Creating Random Numeric MapsSometimes it may be desirable to create maps containing a random value for each data item. This can be accomplished by first creating a map and then using the "random" transform operation to assign a random number to each entry. Note, however, that the transformation will only be applied to assigned entries in the map (plus the default value), so for this to work properly, all the entries in the original map must be assigned.Incorrect way to create random maps
# Create an empty map with default value 0. Note that this map will NOT contain any assigned entries!
MyRandomMap = new Motif Numeric Map(0) # Apply the random transform operation to change the map values to random numbers between 0 and 10. # However, since the map does not contain any assigned entries, only the default value will be changed # and all motifs will thus default to this same number (randomly chosen between 0 and 10) transform MyRandomMap with random(10) Correct way to create random maps
# Create an empty map where all motifs as explicitly assigned the value 0 using the wildcard operator
MyRandomMap = new Motif Numeric Map(*=0) # Apply the random transform operation to change the map values to random numbers between 0 and 10. # Since all entries were previously assigned, each entry is individually transformed into a new random number. transform MyRandomMap with random(10) Modifying Numeric MapsValues in numeric maps can be set explicitly with the set operation or changed relative to the current values with the arithmetic operations increase, decrease, multiply and divide. Numeric Maps can also be transformed with threshold and transform.
# Increase the value of each map entry in Cutoff by 0.1 (including the default value)
increase cutoff by 0.1 # Multiply the value of each map entry in Map1 by its corresponding value in Map2 multiply Map1 by Map2 Sequence Numeric Map
A Sequence Numeric Map is a data object that associates sequences with numeric values. It is a subtype of Numeric Map which again is a subtype of Map.
Creating Sequence Numeric MapsSequence Numeric Maps can be created by explicitly assigning values to named sequences, sequence collections, sequence partition clusters or sequence ranges. (See documentation under Maps and Numeric Maps). In addition, Sequence Numeric Maps can be created based on numeric properties of sequences or on statistics from feature datasets as described below.Creating Sequence Numeric Maps based on sequence propertiesIn the GUI you can create such maps by selecting "Add New ⇒ Sequence Numeric Map" from the "Data" menu and then go to the "From Property" tab and select the property from the drop-down menu. All numeric properties can be selected, both standard and user-defined.The general syntax for creating Sequence Numeric Maps based on properties in protocols is the following:
SequenceNumericPropertyMap = new Sequence Numeric Map(Property: <propertyName> )
Examples
# Create a numeric map based on the size of each sequence
SequenceNumericMap1 = new Sequence Numeric Map(Property:length) # Create a numeric map based on the user-defined sequence property "geneExpression" SequenceNumericMap2 = new Sequence Numeric Map(Property:geneExpression) Creating Sequence Numeric Maps based on feature track statisticsIn the GUI you can create such maps by selecting "Add New ⇒ Sequence Numeric Map" from the "Data" menu and then go to the "From Statistic" tab. Press the "Select" button to bring up a second popup dialog where you can define the statistic function by selecting the feature track, type of statistic function and any conditions that may limit the function. Press "OK" and "OK" again to create the map.The general syntax for creating Sequence Numeric Maps based on feature track statistics in protocols is the following:
SequenceNumericPropertyMap = new Sequence Numeric Map(Statistic: <statistic function> )
Examples
# Create a numeric map based on the GC-content of each sequence
SequenceNumericMap1 = new Sequence Numeric Map(Statistic:"GC-content" in DNA) # Create a numeric map based on the number of TFBS regions in each sequence SequenceNumericMap2 = new Sequence Numeric Map(Statistic:"region count" in TFBS) # Create a numeric map based on average conservation score of each sequence SequenceNumericMap3 = new Sequence Numeric Map(Statistic:"average value" in Conservation) Motif Numeric Map
A Motif Numeric Map is a data object that associates motifs with numeric values. It is a subtype of Numeric Map which again is a subtype of Map.
Creating Motif Numeric MapsMotif Numeric Maps can be created by explicitly assigning values to named motifs, motif collections, motif partition clusters or motif ranges. (See documentation under Maps and Numeric Maps). In addition, Motif Numeric Maps can be created based on numeric properties of motifs or on motif occurrences in a motif track as described below.Creating Motif Numeric Maps based on motif propertiesIn the GUI you can create such maps by selecting "Add New ⇒ Motif Numeric Map" from the "Data" menu and then go to the "From Property" tab and select the property from the drop-down menu. All numeric properties can be selected, both standard and user-defined.The general syntax for creating Motif Numeric Maps based on properties in protocols is the following:
MotifNumericPropertyMap = new Motif Numeric Map(Property: <propertyName> )
Examples
# Create a numeric map based on the size of each motif
MotifNumericMap1 = new Motif Numeric Map(Property:Size) # Create a numeric map based on the information content of each motif MotifNumericMap2 = new Motif Numeric Map(Property:IC-length) Creating Motif Numeric Maps based on motif occurrencesIn the GUI you can create such maps by selecting "Add New ⇒ Motif Numeric Map" from the "Data" menu and then go to the "From Track" tab. Select the motif track in the top-most drop-down menu and the statistical function from the "Property" menu. Available statistical functions are:
The general syntax for creating Motif Numeric Maps based on motif occurrences in a motif track is the following:
MotifOccurrencesMap = new Motif Numeric Map(Track:<motif track name>, property=<statistical function>
, Sequence Collection=<subset>, within=<region dataset> )
Examples
# Create a numeric map based on the total number of occurrences of each motif in the TFBS track
MotifOccurrences = new Motif Numeric Map(Track:TFBS, property=Total count) # Create a numeric map based on the number of sequences that contain an occurrence of the motif within a repeat region MotifOccurrences = new Motif Numeric Map(Track:TFBS, property=Sequence support, within=RepeatMasker) # Create a numeric map with the frequency of each motif within sequences in the Downregulated collection MotifOccurrences = new Motif Numeric Map(Track:TFBS, property=Frequency, Sequence Collection=Downregulated) Module Numeric Map
A Module Numeric Map is a data object that associates modules with numeric values. It is a subtype of Numeric Map which again is a subtype of Map.
Creating Module Numeric MapsModule Numeric Maps can be created by explicitly assigning values to named modules, module collections, module partition clusters or module ranges. (See documentation under Maps and Numeric Maps). In addition, Module Numeric Maps can be created based on numeric properties of modules or on module occurrences in a module track as described below.Creating Module Numeric Maps based on module propertiesIn the GUI you can create such maps by selecting "Add New ⇒ Module Numeric Map" from the "Data" menu and then go to the "From Property" tab and select the property from the drop-down menu. All numeric properties can be selected, both standard and user-defined.The general syntax for creating Module Numeric Maps based on properties in protocols is the following:
ModuleNumericPropertyMap = new Module Numeric Map(Property: <propertyName> )
Examples
# Create a numeric map based on the number of component motifs in each module
ModuleNumericMap1 = new Module Numeric Map(Property:Cardinality) # Create a numeric map based on the sum of the IC-contents for the motif models # having the highest IC for each component motif ModuleNumericMap2 = new Module Numeric Map(Property:Max IC) Creating Module Numeric Maps based on module occurrencesIn the GUI you can create such maps by selecting "Add New ⇒ Module Numeric Map" from the "Data" menu and then go to the "From Track" tab. Select the module track in the top-most drop-down menu and the statistical function from the "Property" menu. Available statistical functions are:
The general syntax for creating Module Numeric Maps based on module occurrences in a module track is the following:
ModuleOccurrencesMap = new Module Numeric Map(Track:<module track name>, property=<statistical function>
, Sequence Collection=<subset>, within=<region dataset> )
Examples
# Create a numeric map based on the total number of occurrences of each module in the CRM track
ModuleOccurrences = new Module Numeric Map(Track:CRM, property=Total count) # Create a numeric map based on the number of sequences that contain an occurrence of the module within a repeat region ModuleOccurrences = new Module Numeric Map(Track:CRM, property=Sequence support, within=RepeatMasker) Text Maps
A text map is a subtype of the general Map type where the values associated with each data item are text strings.
There are three different types of text maps – Sequence Map, Motif Map and Module Map
– that hold values for sequences, motifs and modules, respectively. (Note that the names of these types do not include "Text Map" just "Map").
Text maps that contain "Data⇔Value" associations for other types of data can be defined using Text Variables.
Creating Text MapsThe general syntax for creating Text Maps in a protocol is shown below.
MyTextMap = new <Type> Map(<key1>=<value1>, <key2>=<value2>, ..., <keyN>=<valueN>, _DEFAULT_=<value>)
The argument is a comma-separated list of "key=value" pairs where the keys can be the name of a single data object (of the applicable type) or a collection. The wildcard operator (*) is also supported. In MotifLab v2, the key can also be a reference to a partition cluster, or it can refer to a range of data objects. The value on the right-hand side of an assignment can be any text string but may in some cases require special formatting. If you want the value of a key-value pair itself to contains commas, the whole value must be enclosed in double quotes so that these commas can be discerned from the other commas separating the key-value pairs. Double quotes can optionally be used around all values, even those that do not contain commas. A value can contain internal quotes as long as these are properly opened and closed. If you want to include double quotes inside a value that is already surrounded by quotes, the internal quotes must be escaped with a backslash-prefix, like so: \" ExamplesConsider the following Text Map that contains two entries with "complicated" values. The value for the second entry contains a comma, whereas the third entry value contains quotes. The first set of examples below illustrate proper ways to create this map, while the second set of examples will not work.
Correct ways to create the map above
# The value for the second entry is correctly enclosed with quotes. The third entry can actually be left as is.
MyTextMap = new Motif Map(MM0001=E-box, MM0002="Fos,Jun", MM0003=A "CREB-like" motif) # Here, all the values are surrounded by quotes, but the internal quotes in the third value must then be escaped MyTextMap = new Motif Map(MM0001="E-box", MM0002="Fos,Jun", MM0003="A \"CREB-like\" motif") Incorrect ways to create the map above
# The value for the second entry is not enclosed in quotes, so MotifLab believes that the comma between Fos and Jun
# separates successive key-value pairs. This will lead to an error since "Jun" is not a properly formatted pair. MyTextMap = new Motif Map(MM0001=E-box, MM0002=Fos,Jun, MM0003=A "CREB-like" motif) # Here, all the values are surrounded by quotes, but the internal quotes in the third value causes problems # since they are not properly escaped MyTextMap = new Motif Map(MM0001="E-box", MM0002="Fos,Jun", MM0003="A "CREB-like" motif") Modifying Text MapsValues in text maps can be assigned explicitly with the set operation. Text Maps can also be changed relative to their current values with the arithmetic operations increase, decrease, multiply and divide, but note that these behave differently when applied to Text Maps compared to Numeric maps. The increase and multiply operations both behave like "set union" operators that will add new entries to a comma-separated value list (unless the list already contains the entries). Decrease and divide, on the other hand, behave like "set minus" operators that will remove entries from a comma-separated value list.The threshold and transform operations are not supported for Text Maps. Examples
# Create a few initial data objects
Map1 = new Motif Map(MM0001="A", MM0002="B", MM0003="C") Map2 = new Motif Map(MM0001="X", MM0002="Y", MM0003="Z,W") Col1 = new Motif Collection(MM0001,MM0002) Text1 = new Text Variable("B") Text2 = new Text Variable("A,R") # Now start manipulating Map1 by adding the entry "R" to each map value increase Map1 by "R" # The values in Map1 are now: MM0001="A,R" and MM0002="B,R" and MM0003="C,R" increase Map1 by Text1 in collection Col1 # The value of Text1, which is "B", is added to the map entries for MM0001 and MM0002 (as members of Col1) # However, since the list-value for MM0002 already contains "B" it will not be added a second time. # The values in Map1 are now: MM0001="A,R,B" and MM0002="B,R" and MM0003="C,R" multiply Map1 by Map2 # "multiply" has the same effect as "increase" and will add the corresponding values from Map2 to the lists in Map1. # The values in Map1 are now: MM0001="A,R,B,X" and MM0002="B,R,Y" and MM0003="C,R,Z,W" decrease Map1 by "B" # Decrease will remove entries from the lists # The values in Map1 are now: MM0001="A,R,X" and MM0002="R,Y" and MM0003="C,R,Z,W" decrease Map1 by "A,R" # When a constant string value is used as argument, the value is interpreted as a single value rather than a list # of individual values. Since none of the list-entries in the maps correspond directly to 'A,R', the map will not be # changed and the values in Map1 are still: MM0001="A,R,X" and MM0002="R,Y" and MM0003="C,R,Z,W" # Note that this also applies to increase. If you increase by "A,R" this will be added as a single value # which just happens to include a comma. (In the GUI such values will be enclosed in brackets). decrease Map1 by Text2 # However, if the argument is another data object rather than a string constant, it will be interpreted as a list. # So in this case the two values "A" and "R" (defined by Text2) will be removed from the lists # The values in Map1 are now: MM0001="B,X" and MM0002="B,Y" and MM0003="C,Z,W" # Note that the operations will also be applied to the default value unless limited to a collection. # Since the default value of Map1 was not specified, it started out as an empty string but was changed to "R" # by the first increase operation. This value persisted until the "R" was removed by the last decrease operation here. Sequence Map
A Sequence Map (also called Sequence Text Map) is a data object that associates sequences with textual values.
It is a subtype of Text Map which again is a subtype of Map.
Creating Sequence MapsSequence Maps can be created by explicitly assigning values to named sequences, sequence collections, sequence partition clusters or sequence ranges. (See documentation under Maps and Text Maps). In addition, Sequence Maps can be created based on properties of sequences as described below.Creating Sequence Maps based on sequence propertiesIn the GUI you can create such maps by selecting "Add New ⇒ Sequence Map" from the "Data" menu and then go to the "From Property" tab and select the property from the drop-down menu. All properties can be selected, both standard and user-defined, textual or numeric. (Note that numeric values will be converted to text strings in the map.)The general syntax for creating Sequence Maps based on properties in protocols is the following:
SequencePropertyMap = new Sequence Map(Property: <propertyName> )
Examples
# Create a map based on the chromosome name of each sequence
SequenceMap1 = new Sequence Map(Property:chromosome) # Create a map based on the user-defined sequence property "geneExpression" SequenceMap2 = new Sequence Map(Property:geneExpression) Motif Map
A Motif Map (also called Motif Text Map) is a data object that associates motifs with textual values.
It is a subtype of Text Map which again is a subtype of Map.
Creating Motif MapsMotif Maps can be created by explicitly assigning values to named motifs, motif collections, motif partition clusters or motif ranges. (See documentation under Maps and Text Maps). In addition, Motif Maps can be created based on properties of motifs as described below.Creating Motif Maps based on motif propertiesIn the GUI you can create such maps by selecting "Add New ⇒ Motif Map" from the "Data" menu and then go to the "From Property" tab and select the property from the drop-down menu. All properties can be selected, both standard and user-defined, textual or numeric. (Note that numeric values will be converted to text strings in the map.)The general syntax for creating Motif Maps based on properties in protocols is the following:
MotifPropertyMap = new Motif Map(Property: <propertyName> )
Examples
# Create a map based on the IUPAC consensus for each motif
MotifMap1 = new Motif Map(Property:Consensus) # Create a map based on the names of TFs associated with each motif MotifMap1 = new Motif Map(Property:Factors) Module Map
A Module Map (also called Module Text Map) is a data object that associates modules with textual values.
It is a subtype of Text Map which again is a subtype of Map.
Creating Module MapsModule Maps can be created by explicitly assigning values to named modules, module collections, module partition clusters or module ranges. (See documentation under Maps and Text Maps). In addition, Module Maps can be created based on properties of modules as described below.Creating Module Maps based on module propertiesIn the GUI you can create such maps by selecting "Add New ⇒ Module Map" from the "Data" menu and then go to the "From Property" tab and select the property from the drop-down menu. All properties can be selected, both standard and user-defined, textual or numeric. (Note that numeric values will be converted to text strings in the map.)The general syntax for creating Module Maps based on properties in protocols is the following:
ModulePropertyMap = new Module Map(Property: <propertyName> )
Examples
# Create a map based on the names of the constituent motifs for each module
ModuleMap1 = new Module Map(Property:Motifs) Numeric Variable
The Numeric Variable is the simplest data type in MotifLab and can only hold a single numeric value.
New numeric variables can be created with the operation new (using either a literal number,
a collection, or a numeric map as argument) or by extracting numeric values from analyses or other data objects.
Numeric Variables can also be manipulated with arithmetic operations (increase, decrease, multiply and divide)
and transform.
Creating Numeric Variables
# Create a new Numeric Variable with the specific value 3.14
NumericVar1 = new Numeric Variable(3.14) # Create a new Numeric Variable based on the number of entries in the given collection NumericVar2 = new Numeric Variable(SequenceCollection1) # Create a new Numeric Variable based on the default value from the given Numeric Map NumericVar3 = new Numeric Variable(NumericMap2) # Create a new Numeric Variable by extracting the largest value from a given Numeric Map NumericVar4 = extract "top value" from NumericMap2 as Numeric Variable # Create a new Numeric Variable by extracting a specific result from an analysis NumericVar5 = extract "p-value at least observed overlap" from Analysis1 as Numeric Variable # Create a new Numeric Variable derived from the value of another Numeric Variable NumericVar6 = multiply NumericVar1 by 10 Using Numeric VariablesNumeric Variables can be used to represent numeric values wherever they may appear, such as in arguments for other operations.
# Create a new Numeric Variable named "Cutoff" and use this as an argument in the subsequent
# filter operation to delete regions that have a score below this value Cutoff = new Numeric Variable(90) filter RegionDataset1 where region score < Cutoff Text Variable
The Text Variable is one of the structurally simplest data types in MotifLab (second only to Numeric Variable),
but since it can contain basically any form of textual information it is also one of the most versatile. It can often be used as a general substitute for other more complex types such as collections,
partitions and maps. The data contained in a Text Variable is either a single text string or multiple lines of text,
and depending on how the text is organized, Text Variables can be treated as either lists, sets, tables or documents (in some specific format or just free text).
Creating Text Variables
# Create a new Text Variable with a single line of text
TextVar1 = new Text Variable("a single line of text") # Escape double quotes with \" and TABs with \t TextVar2 = new Text Variable("first column\tsecond \"quote\" column") # Multiple lines of text should be defined as a comma-separated list TextVar3 = new Text Variable("the first line","the second line") # Read contents from file TextVar4 = new Text Variable(File:"dir/subdir/filename") # Read contents from an Output Data object TextVar5 = new Text Variable(Input:Output1) Using Text VariablesText variables can be used to hold information in free-text or structured formats, they can provide textual values for operation arguments or function as substitutes for general collections, partitions or maps. They can also serve as templates for configurable output formats such as Template, TemplateHTML and Properties.
# The Text Variable defines the name of a repeat region type and is used with the filter operation
# to remove only repeats of this type from the track TextVariable1 = new Text Variable("Alu") filter RepeatMasker where region's type equals TextVariable1 # Here the Text Variable represents a collection of repeat region types and will filter repeats of # any of the three types: AluJo, MIR and L2 TextVariable2 = new Text Variable("AluJo","MIR","L2") filter RepeatMasker where region's type is in TextVariable2 # The RepeatClass Text Variable below is formatted as a map containing "key=>value" pairs. # When this map is used in conjunction with the replace operation on the RepeatMasker region dataset # the type property of every region that matches a key in the map will be replaced by its corresponding # value. The result here being that all "Alu" and "MIR" regions are renamed to "SINE" and all "L1" and "L2" # regions are renamed to "LINE" RepeatClass = new Text Variable("Alu=>SINE","MIR=>SINE","L1=>LINE","L2=>LINE") replace RepeatClass with RepeatClass in RepeatMasker # In addition to the regular TFBS track and collection of discovered motifs, the motif discovery method # below returns an additional file with some extra information about the results. Since this information # is not structured in any way that could be suitably represented by another data type,such as a Motif Map, # the information is simply stored in free-text in a Text Variable object (here called "ExtraInfo"). [TFBS,Motifs,ExtraInfo] = motifDiscovery in DNA with MDmethod { ... } # In this example, information about gene expression is read from a file into a Sequence Numeric Map. # The upregulated genes with positive fold-change are stored in a collection, and the largest positive # fold-change value is also extracted from the map. The number of upregulated genes along with the most # extreme fold-change value are reported in custom output format using a template stored in a Text Variable. # Notice how the template text refers to the data objects by enclosing their names in curly braces. GeneExpression = new Sequence Numeric Map(File:"...") UpregulatedGenes = new Sequence Collection(Map:GeneExpression>0) HighestValue = extract "top value" from GeneExpression as Numeric Variable TemplateText = new Text Variable("{UpregulatedGenes:size} genes were upregulated with maximum fold-change {HighestValue}") Output1 = output TemplateText in Template format Text manipulationMotifLab v2 introduced several new ways to manipulate the contents of a Text Variable with the extract and replace operations.Replace or add text The "replace" operation can be used to replace parts of the text matching a regular expression with a new text or to add new lines to the beginning or end of the Text Variable
# Searches for the given text in the Text Variable and replaces every matching instance with a new text
replace "search expression" with "replacement text" in TextVariable1 # Adds a new line of text to the beginning of the Text Variable. # The text can contain \t for TABs or \n to split it over multiple lines replace beginning with "new line of text" in TextVariable1 # Adds a new line of text to the end of the Text Variable. # The text can contain \t for TABs or \n to split it over multiple lines replace end with "new line of text" in TextVariable1 List operations The following extract-functions treat the Text Variable as an ordered list of elements (lines) that could possibly contain duplicate entries.
# Sorts the lines of List1 according to a natural sort order
List2 = extract "sorted" from List1 as Text Variable # Reverses the order of the lines in List1 List2 = extract "reverse" from List1 as Text Variable # Returns a new list where duplicate lines in the original have been removed # so that all entries in the new list are now unique List2 = extract "unique" from List1 as Text Variable # Returns a list containing only those elements that occur multiple times in List1 # (each duplicate is only listed once in the new list) List2 = extract "duplicates" from List1 as Text Variable # Takes all the lines from List2 and adds them to the end of List1 List3 = extract "append:List2" from List1 as Text Variable # Returns only those lines from List1 that contain the specified search text (or not) List2 = extract "lines containing:<text>" from List1 as Text Variable List2 = extract "lines not containing:<text>" from List1 as Text Variable # Returns only those lines from List1 that match the specified regular expression (or not) # Note that the expression must match the full line, not just parts of it, so if you want to # search for text that could occur anywhere within a line you must begin and end the regex with ".*" List2 = extract "lines matching:<regex>" from List1 as Text Variable List2 = extract "lines not matching:<regex>" from List1 as Text Variable Set operations
These extract-functions treat Text Variables as a mathematical sets (member collections), or rather as a cross between a set and a list.
# Finds all elements from Set2 that are not already present in Set1 and adds them to the end of Set1
Set3 = extract "union:Set2" from Set1 as Text Variable # Removes all elements from Set1 that are also present in Set2 Set3 = extract "subtract:Set2" from Set1 as Text Variable # Removes all elements from Set1 that are not present in Set2 Set3 = extract "intersect:Set2" from Set1 as Text Variable # Finds all elements from Set2 that are not already present in Set1 and adds them to the end of Set1. # However, elements that are present in both sets will be removed from the result. # If Set2 contains duplicates not found in Set1, these will be added as duplicates. Set3 = extract "xor:Set2" from Set1 as Text Variable Table operations
These extract-functions treat the Text Variable as a table with each line representing a row and with columns separated by TABs.
# Transposes Table1 so that the original rows becomes columns in the new table and vice versa
Table2 = extract "transpose" from Table1 as Text Variable # Creates a new table based on columns 2, 4 and 5 from Table1 (assuming it has at least 5 columns) Table2 = extract "columns: 2,4,5" from Table1 as Text Variable # Creates a new table based on columns 1 and 2 from Table1, followed by columns 3 through 5 # and then the last three columns (end-2, end-1 and end). Table2 = extract "columns: 1,2,3:5,end-2:end" from Table1 as Text Variable # Reverses the order of all the columns in Table1 Table2 = extract "columns: end:1" from Table1 as Text Variable # Creates a new table based on columns 1, 6, 5 and 4 from Table1, # then a new column containing the value "1000" in all rows and finally column 1 is repeated once more. Table2 = extract "columns: 1,6:4,'1000',1" from Table1 as Text Variable It is also possible to create a table by concatenating columns from multiple Text Variables using the new operation.
# Creates a new Text Variable table with three columns based on the specified Text Variables
Table1 = new Text Variable(columns:TextVar1, TextVar2, TextVar3) Background Model
Background models define probability distributions for DNA sequences. These can be simple (0-order) models that only contain information about the relative frequencies of the four DNA bases in the sequence
or they can be higher-order Markov models (up to order 5)
where the probability of observing a particular base at a position in a sequence will depend on which bases that preceeded it.
For a Markov model of order N the Background Model will store information about the relative frequency of every oligo of length N (of which there are 4N)
and also a transition matrix of size 4N×4 which states the probabilities that a given oligo of length N will be followed by either an A, C, G or T respectively.
In addition, the model will also contain information about the single nucleotide frequency of each of the four DNA bases.
Creating Background ModelsBackground models can be defined manually by explicitly listing all the oligo frequencies and transition probabilities, but this is not recommended for higher-order models since it would involve too much tedious typing that can be hard to do correctly. A better way to create background models is to derive them from DNA sequence tracks. MotifLab also comes bundled with several predefined background models (borrowed from the INCLUSive project) that can be easily imported, and background models can be imported from files in various formats.
# Import the predefined "EDP_human_3" background model that comes bundled with MotifLab
EDPhuman3 = new Background Model(Model:EDP_human_3) # Import a background model from file in MEME background format BGmodel = new Background Model(File:"C:\mouse.freq", Format=MEME_Background) # Create a new 0-order model with uniform distribution UniformBG = new Background Model # Manually define a 0-order model with high GC-content (A=10%, C=40%, G=40%, T=10%) High_GC_background = new Background Model(SNF:0.1,0.4,0.4,0.1;MATRIX:0.1,0.4,0.4,0.1) # Create a new 3-order background model derived from a DNA track. # Use the DNA strand relative to the sequence orientation Background1 = new Background Model(Track:DNA, Order=3, Strand=Relative) Modifying Background ModelsBackground models are immutable data objects and cannot be changed after they have been created.Using Background ModelsBackground models can be used by some motif discovery and motif scanning tools to correct for background bias when searching for transcription factor binding sites. Background models can also be used to create new artifical DNA sequence tracks or mask portions of existing DNA tracks.
# Use the "EDP_human_3" model to correct for background bias when discovering motifs with MEME
[TFBS,MEMEmotifs] = motifDiscovery in DNA with MEME {Background=EDP_human_3, ... } # Replace bases inside TFBS regions with new bases randomly sampled from the distribution defined # in "Background1". This will in effect destroy these binding motifs in the sequence. mask DNA with Background1 where inside TFBS # Create a new artificial DNA sequence track by randomly sampling bases # according to the distribution defined in the background model DNA_random = new DNA Sequence Dataset(EDP_human_3) Expression Profile
An Expression Profile is a data table primarily meant to hold gene expression profiles, wherein each sequence (gene) can be associated with multiple numeric values for different conditions.
It can be thought of as a two-dimensional extension of the Sequence Numeric Map, where each column in the table corresponds to a SNM.
The columns of the table are referred to as conditions (or sometimes experiments). By default, the conditions are numbered (starting at 1), but they can also be given explicit names.
It is possible for some columns to have names and others to just have default numbers, but it is not recommended to mix these styles (especially since you can give a column a number as an explicit name, which can be confusing).
Creating Expression ProfilesExpression Profiles can be created manually, either by explicitly assigning condition values to sequences, by basing the profile on a list of Sequence Numeric Maps, or by basing it on a subset of conditions from another existing profile.To create an expression profile in the GUI, select "Add New ⇒ Expression Profile" from the "Data" menu or press the plus-button in the Data Objects Panel and select "Expression Profile" from the context menu. You can then edit the values in the table. The new profile will start off with only one column, but you can add more by pressing the "Add condition" button in the dialog. To rename a condition, right-click on the column header and select "Rename" from the context-menu. In a protocol, you can create a profile by assigning a comma-separated list of values to each sequence. Each sequence must have the same number of values, and the sequence entries should be separated by semicolons. Existing sequences that are not included in the list will have their values set to 0 for all conditions by default. You can assign names to the conditions by including entries on the form "header[column number]=<name>;". Examples
# Create an expression profile with four conditions (numbered 1, 2, 3 and 4)
# Only the two listed sequences will have explicitly assigned values, and all other sequences # will default to zero for all conditions ExpressionProfile1 = new Expression Profile(ENSG00000100345=2.4,0.3,-0.2,0.5;ENSG00000111249=1.8,-0.4,0.1,1.2) # Same as above, but now the second and fourth columns/conditions are given explicit names, # so the conditions will now be named: 1, Case, 3, Control ExpressionProfile2 = new Expression Profile(header[2]=Case;header[4]=Control; ENSG00000100345=2.4,0.3,-0.2,0.5;ENSG00000111249=1.8,-0.4,0.1,1.2) Creating Expression Profiles from a list of Sequence Numeric MapsIn a protocol, Expression Profiles can be created from a comma-separated list of Sequence Numeric Maps. The names of the conditions/columns will be based on the names of the maps.
# Create a profile based on the three maps. The columns will be called: Sample1, Sample2 and Sample3
ExpressionProfile3 = new Expression Profile(Map:Sample1, Sample2, Sample3) Creating Expression Profiles based on a subset of another profileA new Expression Profile can be created by cherry-picking conditions from another profile. This is accomplished with the extract operation using the "subprofile:" property. You can extract a comma-separated list of named or numbered columns, and it is also possible to define column ranges on the format "firstColumn-lastColumn" (or "firstColumn:lastColumn"). If the last column in a range is located before the first column in the original profile table, the order of the columns in the range will be reversed in the final profile. Columns that have explicitly assigned names (even if these names are numbers) in the original profile will retain their names in the new profile as well, but columns that just have default column numbers will not retain these numbers. So if you extract columns "3,5,6" from a profile with just default numbered columns, they will be numbered "1,2,3" in the new profile. Note that a column with an explicit name can not be added twice to a second profile.Examples
# Create a new profile based on the first four columns of Profile1 (assuming default numbering)
Profile2 = extract "subprofile:1,2,3,4" from Profile1 as Expression Profile # Another way to create a new profile based on the first four columns of Profile1 using a column range Profile3 = extract "subprofile:1-4" from Profile1 as Expression Profile # Create a new profile based on the first four columns of Profile1 but in reversed order Profile4 = extract "subprofile:4-1" from Profile1 as Expression Profile # Create a new profile based on the column named "Case" followed by columns 3 through 5 and then the column "Control" Profile5 = extract "subprofile:Case,3-5,Control" from Profile1 as Expression Profile Importing Expression Profiles from filesYou can import an expression profile from file by selecting "Import Data..." from the "Data" menu and then choosing "Expression Profile" from the type drop-down menu in the dialog, or by pressing the plus-button in the Data Objects Panel, selecting "Expression Profile" from the context menu and then going to the "Import" tab in the dialog. Three data formats are currently supported for expression profiles: ExpressionProfile, ExcelProfile and Plain. Note that condition names are not supported by the Plain format.Modifying Expression ProfilesSimilar to Numeric Maps, values in Expression Profiles can be set explicitly with the set operation or changed relative to the current values with the arithmetic operations increase, decrease, multiply and divide. Expression Profiles can also be transformed with threshold and transform. Note that, like Numeric Maps, these operations can be limited to a subset of the sequences in the profile, but it is currently not possible to limit the application of the operation to a subset of conditions. Hence, when modifying the profile, all the values for a sequence will be updated. It is also not currently possible to modify a profile using a second profile, for instance to subtract the values in one profile from the values of second profile.
# Increase all the values in the expression profile by 0.2
increase ExpressionProfile1 by 0.2 # Multiply the gene expression values for each sequence by its corresponding value in the map # (all values for the same sequence will be multiplied by the same value across all conditions in the profile) multiply ExpressionProfile2 by SequenceNumericMap1 # Set all the profile values to 0 for sequences in the Downregulated set (across all conditions) set ExpressionProfile1 to 0 in collection Downregulated Using Expression ProfilesThe main use of Expression Profiles is as additional input to motif discovery programs or other external programs that can use this type of information to improve their analysis. However, so far no such programs are supported.Priors Generator
A Priors Generator (PG) is an object that can estimate an a priori probability that a position in a sequence could be associated with a specific feature.
This estimate is usually based on the values of other feature tracks that correlate with the target feature.
PG objects can be used to create position-specific priors tracks (aka "positional priors") that display a prior probability of finding a certain feature, such as TF binding sites, at each position in a sequence.
When such tracks are used as input to e.g. motif discovery or motif scanning tools,
they can guide the tools to the parts of the sequence that are more likely to contain the target feature and thereby help them make better predictions.
Creating Priors GeneratorsPriors Generators are usually trained by supervised machine learning methods to predict the presence of a target feature based on a set of input features, for instance predicting the possible presence of TF binding sites based on features such as sequence conservation, DNase hypersensitivity, and various histone marks (H3K4me3, H3K4me1, etc.)The MotifLab GUI includes a "wizard" to train a PG in a few steps, but in order to do that you need to have a training dataset with annotated regions of the feature you want to predict as well as access to additional feature tracks that can be used as input to predict the target feature. To create a new PG, select "Add New ⇒ Priors Generator" from the "Data" menu or press the plus-button in the Data Objects panel and select "Priors Generator" from the drop-down menu. The Priors Generator dialog will let you import a finished, pre-trained PG from a file, to re-train a new PG based on predefined configuration or to create a completely new PG in 3 steps, as described below. Training a new Priors GeneratorStep 1: Selecting the target features, input feature and classifier The target feature is the feature that you want the Priors Generator to be able to predict. In order to train the PG you must already have a region dataset with correctly annotated regions for that feature, which is selected with the drop-down menu in the top-left part of the dialog. The input features that you will use to predict the target are selected from the list underneath. This list will be populated with all the other region and numeric feature datasets that you currently have. The selected input features should ideally show at least some correlation with the target, either alone or in combination, or else it will be impossible to use them for prediction. The right-hand side of the first panel configures the classifier(s) – the machine learning methods you will use for the prediction. You can add a classifier by first selecting the type from the drop-down menu and then pressing the "Add new" button in front of it. Three types of classifiers are currently included in MotifLab: neural networks, naive Bayes classifiers and decision trees. You can edit the properties of a classifier you have already added (such as the number of hidden layers and nodes in a neural network) by selecting it in the list and pressing the "Edit" button. It is possible add multiple classifiers, and these will then be combined into an ensemble classifier that is trained using an adaptive boosting approach. When you are satisfied with your selections at this step, press the "Next" button to move on to the second step. 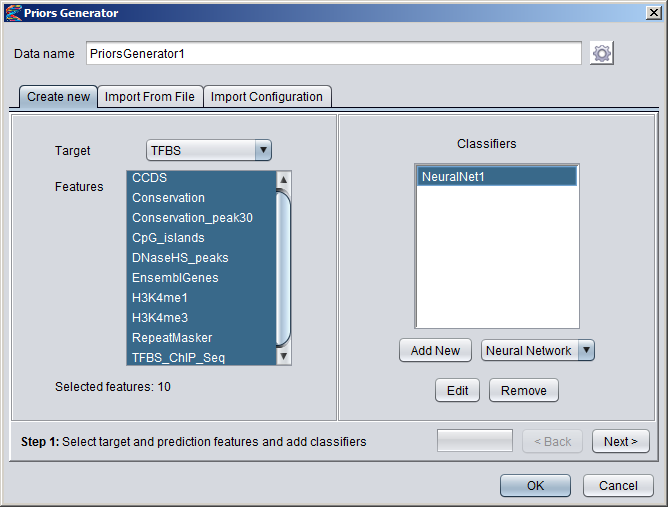
Step 2: Setting up the training and validation datasets The second step involves setting up a dataset that will be used for training the classifier(s) as well as an independent validation dataset that can be used to verify that the classifier(s) are able to generalize well to new cases they have not seen before. Technically, every position in all your sequences could be used as training examples, but since training with a very large dataset is extremely resource demanding, it is wiser to select a smaller subset of positions to use a training examples. The number of positions to sample for the training set is selected in the top-right box, and the sampling strategy is selected in the top-left drop-down menu (and visualized underneath with red segments indicating target feature regions, white is background and the black marks below are sampled positions). The possible ways to sample are:
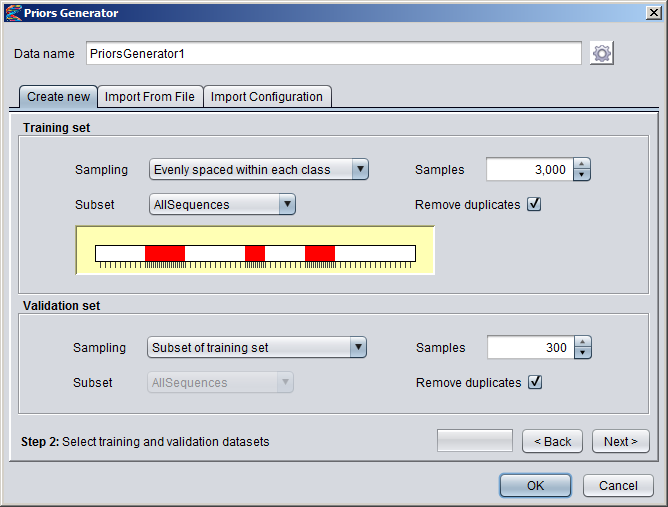
After you have made your selections at this step, press the "Next" button to start the sampling process and create the datasets. When the datasets are finished, MotifLab will display a popup dialog showing you the number of positive and negative examples in each of the datasets. If you are satisfied, press the "Yes" button to move on to the final step, or press "No" to go back and try sampling again. If you like, you can also save the datasets at this point (in Attribute-Relation File Format). 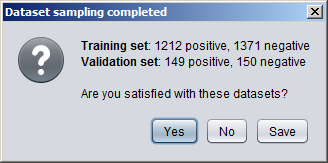
Step 3: Training the classifiers The last step is to train the classifier(s). The only thing you usually need to do here is to press the "Train" button to start the training. If you are training an ensemble with multiple classifiers, the "sampling" setting can control how to adjust the training set for the different classifiers by using "weighted examples". When this option is employed (by selecting either "weighted" or "stochastic universal" sampling), the first classifier will be trained on the full training set, and each example that it classifies correctly will have its associated "weight" reduced. When the next classifier is to be trained, a new training set of the same size is constructed by sampling – with replacement – from the original set in such a way that examples with higher weights have a higher chance or being selected (possibly even multiple times). This means that the new training set will be focused more towards the training examples that the previous classifiers failed to classify correctly. The selection of training examples could be done by repeatedly sampling each example to include at random with a probability proportional to its relative weight ("weighted"), which should on average result in each training example being selected a number of times which is proportional to its weigth, but in extreme cases the selection could be skewed. The "stochastic universal" sampling strategy, on the other hand, only uses one round of sampling and guarantees that each example is included in a number proportional to its weight. For instance, if you have 4 training examples with weights 4.0, 2.0, 1.0 and 1.0, the new dataset should, on average, include 2 copies of the first example (since the weight of this example amounts to 50% of the total weight), one copy of the second example, and one copy of either the third or fourth example. The "stochastic universal" sampling strategy will ensure that this really will be the result, but with the more random "weighted" strategy, you could in theory end up selecting four copies of the last example. If a classifier is trained over multiple iterations, you will be able to see the progress of the classifier over time in the top graph, where the blue line shows the performance on the training set and the red line on the validation set. The performance is measured by accuracy, i.e. the fraction of correctly classified examples (ie. (TP+TN)/(TP+TN+FP+FN) ). This fraction is also showed in the table and beneath the pie charts. If the classifier is not refined over multiple iterations, you will only see the final performance after the training process is finished. The two pie charts show the results on the training and validation sets. The green colors indicate examples that were correctly classified, whereas the red indicate the proportion of examples that were misclassified. The darker (more saturated) colors represent the positive examples (target feature) whether these were correctly classified (green) or not (red), whereas the lighter colors represent the negative examples (background) that were either correctly (light green) or incorrectly classified (light red). Or in other words: Dark green = True Positives (TP), Light green = True Negatives (TN), Light red = False Positives (FP) and Dark red = False Negatives (FN). 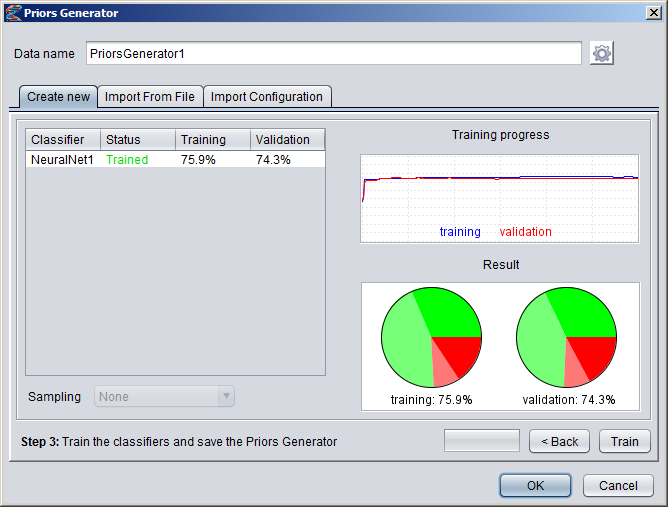
When the training process is finished, the "Train" button will turn into a "Save" button, and you will have to save the Priors Generator before you can finish the process. The main reason for this is that Priors Generators are too complex to describe in a single line in a protocol, so the protocol has to save all the required information to a separate file that it can then reference. The PG can be saved either "as is" in its current trained form, or you can save a configuration file describing all the selections you have made in the two first steps (example). MotifLab will then re-train a new PG on-the-fly as needed based on this configuration setup when the protocol is executed. If you are not satisfied with the final performance of the PG and want to try training it again (before saving), you can press the "<Back" button once and the "Save" button will then go back to being a "Train" button again. If you press "<Back" a second time, you will be taken back to the previous step. Initializing Priors Generators in a protocol
# Importing a pre-trained Priors Generator from file
PriorsGenerator1 = new Priors Generator {File:"C:\MotifLab\PG_for_predicting_TFBS.pge", format=PriorsGeneratorFormat } # Importing a configuration file describing how to create a Priors Generator # After importing the file, MotifLab will train a new PG on-the-fly based on the description PriorsGenerator2 = new Priors Generator {Configuration:"C:\MotifLab\PriorsGenerator_config.xml"} When a Priors Generator is created from a configuration file in a protocol, the configuration file describes how to sample the training and validation datasets. However, it is possible to override these dataset settings individually by addings extra parameters in "parameterName=value" format. The parameter name should be on the form "dataset.property", where dataset is either "trainingset" or "validationset" and property is one of the following:
Example with dataset overrides (See also this example)
# Importing a configuration file describing how to create a Priors Generator
# Irrespective of what is stated in the configuration file, the number of samples used for the training set # will be 2000 and duplicate examples will not be removed PriorsGenerator2 = new Priors Generator {Configuration:"C:\MotifLab\PriorsGenerator_config.xml", trainingset.samples=2000, remove_duplicates=true } Once a PG has been created, you can inspect it by double-clicking on the PG in the Data Objects panel. The figure below shows a Priors Generator based on a single neural network classifier that was trained to predict a track with "TFBS" regions based on 10 different input features (listed in the panel on the left). At each sequence position, the values from these feature tracks will be used as input to the nodes in the top layer (blue). The information provided by these feature values will then be processed by the network (which here has a single hidden layer with 8 nodes) before a final probability value is output by the single node at the bottom of the network. 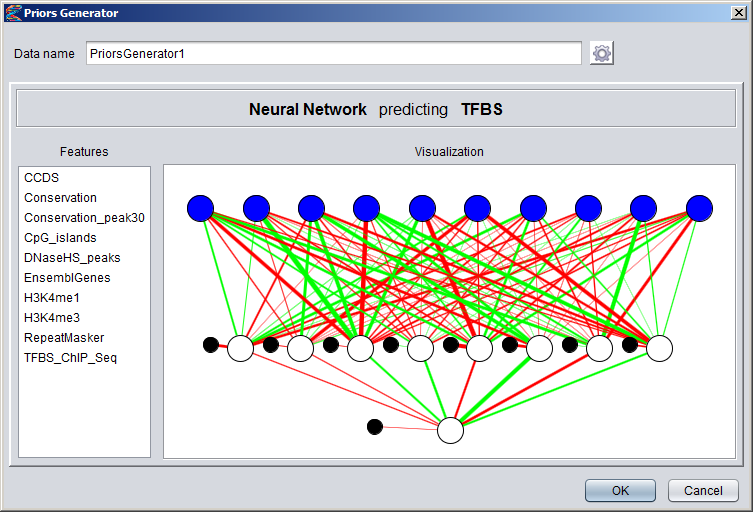
Using Priors GeneratorsPriors Generators can be used by the predict operation to create tracks with positional priors. If the PG was trained to create priors by combining information from several input tracks, these tracks (with the same names but not necessarily from the same sequence locations) must also be available when running the predict operation. (However, the target track used during training is not needed for this step).Analysis
An analysis is a complex data object containing results produced by the analyze operation. Different types of analyses will produce different subtypes of Analysis objects.
Creating AnalysesAn analysis can only be created as output by the analyze operation or returned by some external programs. For some types of compatible analyses, information can be extracted from several analyses and combined into a new analysis using the collate operation.Examples
# Analyzes the GC-content of a DNA track and returns the value for each sequence as well as summary statistics
Analysis1 = analyze GC-content {DNA track = DNA } # Analyzes the correlation (Pearson's and Spearman's) between two compatible numeric maps Analysis2 = analyze numeric map correlation {First = Map1, Second = Map2 } # Counts the number of times each TF motif occurs in a TFBS track and compares these numbers to expected frequencies Analysis3 = analyze count motif occurrences {Motif track = TFBS, Motifs = JASPAR, Expected frequencies = FreqMap } # Analyzes the positional distribution of individual motifs in the TFBS track relative to the TSS of each sequence Analysis4 = analyze motif position distribution {Motif track = TFBS, Motifs = JASPAR, Alignment anchor = "TSS" } # Combines two columns of information from Analysis3 (total and p-value) with one column from Analysis4 (Kurtosis) # to create a new an "bigger" analysis object Analysis5 = collate "total" from Analysis3, "p-value" from Analysis3, "Kurtosis" from Analysis4 Modifying analysesAn analysis data object is meant to represent the final output produced at the very end of a processing workflow. As such, it is not designed to be manipulated further.Using analysesThe information contained in Analysis objects can be inspected by researchers and provide evidence for existing hypotheses or perhaps suggest new ones. Analyses can also be output to documents in various formats, e.g. Excel and HTML with graphs and tables, and these figures can be often be included directly in scientific publications. For example, in the MotifLab paper, figures 3 and 5 were produced by the benchmark analysis, whereas figure 4 was produced by the evaluate prior analysis. Figure 6 shows a table with data collated from multiple analyses.Analysis objects can be viewed in the GUI by double-clicking on an Analysis in the Data Objects panel (or right-clicking and selecting "Display ..." from the context menu). This will open a dialog to display the information contained in the analysis object. These displays can often be interactive. For example, some will only display parts of the information, so the user will have to select which parts to view (using e.g. drop-down boxes). Analyses that includes tables can usually be searched or filtered, and analyses that displays results (graphs and/or tables) for various tracks will often only include the tracks that are currently visible in the GUI (i.e. not "hidden"). The user can therefore decide which results to include in the graph by toggling the visibility of the tracks (although usually you will have to close the Analysis dialog and reopen it again to update the graphs). Output Data
An Output Data object is a document in some specific data format.
It is mostly just used to represent the information held by other data objects in text based formats that can later be saved to files, since data objects in MotifLab cannot otherwise be saved to files directly (in their internal representation).
Output Data objects can also hold more complex documents containing extensive reports, in for example HTML format, that can contain embedded content, such as e.g. images.
Output Data objects are displayed in separate tabs in the main panel of the GUI, with the tabs themselves showing the names of the data objects. The contents of Output Data objects can be saved to files by selecting either "Save", "Save As..." or "Save All" from the file menu. Unless otherwise specified, the names of the output files will be based on the names of the data objects, and the file suffix will be determined from the data format used when creating the Output Data object. If MotifLab is run in CLI-mode, all Output Data objects created during the execution of the protocol (that have not been explicitly deleted) will automatically be saved to files afterwards, unless the "-no_output" option is specified. In the GUI, individual Output objects can be deleted by clicking on the close icons of their tabs, and you can delete all Output objects by selecting "Close All Output Panels" from the "View" menu or by selecting either "Clear Data ⇒ Other Data" or "Clear All Data" from the "Data" menu. Output Data objects can be of three main types: text ("raw text"), HTML or binary formats. The first two of these can be displayed directly in MotifLab's GUI, but documents in binary formats (e.g. Excel formats or PDF) cannot be displayed by MotifLab. However, binary formatted documents can still be saved to files and reopened later in external viewers, such as Excel or Adobe Acrobat. Creating Output DataOutput Data objects can only be created from other data objects using the output operation after selecting which format to use. Objects in most "raw text" formats will allow their documents to be appended to later, but binary and HTML-formatted documents will not.Examples
# Output the contents of a DNA track object (feature dataset) in FASTA format
Output1 = output DNA in FASTA format # If no target Output Data object is specified, the output will be stored in an object called "Results" output DNA in Plain format # First output the contents of a TFBS track object in BED format to the Output Data object "Output2", # then append the contents of a second TFBS track in GFF format to the same document Output2 = output TFBS in FASTA format Output2 = output TFBS2 in GFF format # Output the contents of a DNA track object (feature dataset) in FASTA format Output1 = output DNA in FASTA format Modifying Output DataIf the current contents of an Output Data object allows it, new text can be appended to the end of the document by applying additional output operations that write to the same Output Data object. However, existing parts of a document cannot be modified after they have been added.Using Output DataOutput Data objects can be viewed by the user in MotifLab's GUI. This can be nice for analysis output and similar report-like documents, but the main purpose of most Output Data objects is just to function as temporary storage of another data object in a format that can be saved to file. Like Text Variables, Output Data objects containing text in a supported input data format can also be parsed to create other objects with the new operation.HTML dependenciesOutput Data objects in HTML formats may include embedded content, such as images, CSS style sheets and JavaScript (JS) files, that must also be saved – perhaps to separate files – when the HTML document itself is saved. How this is done is controlled in different ways.CSS and JavaScript The way CSS and JS content is handled can be controlled by selecting "Options..." from the "Configure" menu and then going to the "HTML" tab.
Motif logos (and module logos) Motif logos are graphical representations of the binding preferences for different nucleotide bases each at each position in the TF motif. Many analyses contain tables with results for individual motifs, and when outputting such analyses in HTML format, the logos for these motifs can optionally be included as images. The way this is done is usually decided when executing the output operation using the "Logos" data format parameter.
OperationsIntroductionMotifLab users can apply operations to create, manipulate and analyze data objects. The behaviour of these operations can be controlled with arguments and by specifying conditions to limit their application. Commands to execute different operations can also be combined into protocol scripts that define multi-step workflows.The operations available in MotifLab can broadly be divided into the following groups:
Operation argumentsOperations take arguments that can be used to control the behaviour of the operation. Most operations require a source data argument which specifies the data object(s) the operation should be applied to, and many operations also allow, or even require, a target data argument specifying the (name of the) data object the results from the operation should be stored in. Additional optional or required arguments may have to be provided depending on the particular operations, and applying conditions to an operation will call for specification of still more arguments. For example, the increase operation takes two required arguments: a source argument specifying which data object to increase and a second argument specifying how much to increase the value of the source data object by.The type of values allowed for an argument will depend on the function of that particular argument, but the values can typically be data objects (perhaps limited to specific data types), numeric values or text values. When an argument calls for a numeric value, this can usually be provided as a numeric constant or with a numeric data object such as a Numeric Variable or a Numeric Map. Likewise, if an argument calls for a text value, this can normally be provided as a literal text string (usually enclosed in double quotes) or with a Text Variable or Text Map. Using Maps as argumentsThe use of maps to provide values for arguments in operations warrants some explanation. A map is a data object which describes an association between basic data objects (motif, module or sequence) and their respective values (which can be either numerical or textual depending on the type of map). The map can be thought of as a table with two columns, the first column listing the names of data objects and the second column containing the associated value. In this way it is possible to use the map to look up the value for each data object. When maps are used as arguments, the particular values used by the operation will depend on the "natural context". Consider, for example, the command "increase TFBS by NumericMap1". Assuming that TFBS refers to a motif track (or alternatively a module track), this operation will go through all regions in all sequences in the track and increase the score property of each region by a certain amount which can be found in the map. Now, if NumericMap1 is a Sequence Numeric Map, the value to use for the argument can be found by looking up the name of the "parent sequence" in the map (i.e. the sequence the region belongs to). This means that all the regions belonging to the same sequence will have their scores increased by the same amount, but regions belonging to different sequences could potentially be increased by different values. If, on the other hand, the NumericMap1 argument was a Motif Numeric Map (or Module Numeric Map), then the type property of each region (which in this case should correspond to the name of a Motif or Module) would be used to look up the value to use from the map. The result would be that all regions associated with the same motif/module would have their scores increased by the same amount, even for regions belonging to different sequences. Sometimes it is allowed to use maps in situations where the correct context cannot be properly determined. In these cases the default value of the map will be used. The default map value will also be used if the sequence/motif/module does not have an explicitly assigned value in the map.A note on coordinates and orientationsIn MotifLab, genomic segments can be represented at three different levels that give rise to different coordinate systems and anchor points.
The genome of a species is divided into one or more chromosomes each having a coordinate system starting at position 1. For instance, the 115Mbp long chromosome 13 of the human genome can be described with coordinates "chr13:1-115169878". Sequences are objects representing (sub)segments of chromosomes ranging in size from 1bp up to the full length of the chromosome, although they are mostly used to represent smaller regions of interest, such as e.g. genes or upstream promoter regions of genes. In the figure above, the first sequence ENSG00000035403 represents a segment around the transcription start site of the gene VCL and the second sequence ENSG00000100345 represents a segment around the TSS of gene MYH9. Since VCL is located on the direct strand, the corresponding sequence is also located on the direct strand as indicated by the green arrow pointing towards the right underneath the sequence name. The MYH9 gene, on the other hand, is located on the reverse strand and so the arrow underneath its sequence name points towards the left. Both sequences are visualized here relative to their annotated orientation, which means that the upstream direction in both cases is to the left and the downstream direction is to the right. (It is possible to visualize sequence in either orientation. When sequences are visualized in the opposite orientation of their annotated strand, the arrow underneath the sequence name is shown in red color rather than green). The location of sequences are always stored internally in MotifLab using genomic coordinates relative to the direct strand of the chromosome, and so the "genomic start" of a sequence (also simply called "start") is its smallest genomic coordinate and the "genomic end" of a sequence (or just "end") is its largest genomic coordinate. This applies irrespective of the actual orientation of the sequences. Hence, in the figure above, the "start" of the first sequence is to the left and the "end" is to the right. However, for the second sequence which is located on the reverse strand, the "start" is located to the right and the "end" is on the left. Relative coordinates, on the other hand, are always seen relative to the orientation of the sequence itself, which means that the "relative start" of a sequence is its most upstream coordinate and the "relative end" is its most downstream coordinate. For sequences on the direct strand, the relative coordinates are exactly the same as the genomic coordinates, but for sequences on the reverse strand the "relative start" corresponds to the "genomic end" and the "relative end" corresponds to the "genomic start". To make matters more confusing, sequences can also contain smaller regions that can have their own orientations independent of their parent sequences. The orientation of a TFBS region, for example, will be determined by the orientation of its corresponding binding motif model. When region tracks are visualized with region orientations, the track will be divided into two parts by a horizontal line. Regions in the top half have the same relative orientation as their parent sequence (they are located on the same strand as the sequence), and regions in the bottom half are located on the opposite strand of their parent sequence. For regions, as for sequences, the genomic coordinates "start" and "end" correspond to the smallest and largest genomic coordinates respectively, whereas the relative coordinates are seen relative to the orientation of the parent sequence. Hence, the relative start is always the most upstream coordinate relative to the sequence (left edge of the regions in the figure) and the relative end is the most downstream coordinate (right edge of the regions in the figure). In the region coordinates system, the coordinates are seen relative to the orientation of the regions themselves rather than the orientation of the genome or parent sequence. The "region start" is thus considered to be the first position in the region and the "region end" is the last position within the region. For example, if a region represents the binding sequence "ACAAGT" then the region start is the position of the first base "A" and the region end is the position of the last base "T". (The DNA sequence will depend on the orientation of the parent sequence). In the figure above, the "region start" position corresponds to the left edge of regions in the upper half of the track and to the right edge of regions in the bottom half. ConditionsFeature Conditions
Feature conditions are conditions that can be placed on feature dataset operations
to limit the application of the operation to certain positions (for DNA sequences and numeric datasets)
or regions (for Region datasets). In protocols such conditions are introduced by the keyword "where" following the operation and its arguments.
For example, the following command (without a condition) will apply the mask operation to every position in the DNA sequence and replace every base with the letter "N".
By specifying a condition we can limit the application of the operation to specific parts of the sequence. In the example below the DNA sequence will only be masked inside annotated repeat regions (from the RepeatMasker track).
It is possible to specify multiple conditions for the same operation by connecting them into compound conditions using the boolean operators AND and OR. Position Conditions
The position condition applies to DNA tracks and Numeric tracks and is evaluated for each individual position in a sequence.
Operations will only be applied to positions where the condition holds true. The general syntax for this type of condition in protocols is as follows:
If the optional not keyword is specified immediately after "where", the truth value of the whole condition following it will be inverted. The operand track referred to within the condition itself does not have to be the same as the target track that the operation is applied to, and this operand can be of any type (DNA, Numeric or Region track). Depending on the type of operand track chosen, different comparisons are possible as described in the tables below. Operand track is a Numeric Dataset
The value(s) of Operand2 must be numeric and can be provided either as a constant number, a Numeric Variable, a Sequence Numeric Map or a (second) Numeric Dataset.
If a Sequence Numeric Map is used, the value of Operand2 will be the value for the current sequence in the map.
If a Numeric Dataset is used, the value of Operand2 will be the value in the corresponding position of this track (i.e. the two tracks are compared position by position).
Operand track is a DNA Sequence Dataset
The value of Operand2 must be a single DNA base letter (or IUPAC consensus letter) which can be provided either as a literal string (enclosed in double quotes), a Text Variable or a (second) DNA track.
If the value is provided as a string or Text Variable, only the first letter in the text is used even if the actual string might be longer.
If a second DNA track is used, the value of Operand2 will be the value in the corresponding position of this track (i.e. the two tracks are compared position by position).
By default the comparisons use DNA bases taken from the direct strand (even if the sequence itself has reverse orientation), and this was also the only option available in MotifLab v1. In version 2.0+ of MotifLab, it is possible to use bases from the relative strand by adding the qualifier relative strand after the "equals", "matches" and "case-sensitive equals" operators.
Operand track is a Region Dataset
Illustration: The black regions indicate the bases where the condition holds true for the different comparison operators when the operand is a Region Dataset. 
Examples:
Region Conditions
The region condition applies to Region Datasets and is evaluated for each individual region in a sequence.
Operations will only be applied to regions where the condition holds true. Three different subtypes of region conditions exist.
The first type bases the condition on the value of a specific property of the region itself. The second type compares the region
to other regions from the same or a different region track. The last type bases the condition on the values of a numeric track within
the sequence segment spanned by the region. The general protocol syntax for these three cases are as follows:
The region keyword discriminates this kind of condition from the position condition. For improved language in protocols, the alternative forms region's, regions and regions' are also accepted. If the optional not keyword is specified immediately after "where", the truth value of the whole condition following it will be inverted. Conditions based on region properties
The specification of the property must include its type (text, boolean or numeric) and the name of the property within double quotes. (Boolean properties are treated the same as text properties except that their values are expected to be boolean).
The three standard properties "type" (text), "score" (numeric) and "length" (numeric) are exempt from this rule and can be referred to by the short-hand notation "where region type|score|length ...".
For numeric properties, the value(s) of the operand must be numeric and can be provided either as a constant number, a Numeric Variable or a Numeric Map. If a Motif- or Module Numeric Map is used as the operand, the type property of the region will be used as the key to look up the corresponding value in the map. If a Sequence Numeric Map is used, the value of the operand will be the value for the current sequence in the map. For text properties, the operand can either be a constant string enclosed in double quotes, a Text Variable, a Collection or a Text Map. If a Motif- or Module Map is used as the operand, the type property of the region will be used as the key to look up the corresponding value in the map. If a Sequence Map is used, the value of the operand will be the value for the current sequence in the map. The "equals" and "matches" operators expect the operand to be a single value that must match with the property. The only difference between these two is that "matches" allows the operand to be/contain a regular expression (as explained here). Note that the property value is expected to match the full regular expression, so if you want to target regions where the property contains some specific substring (e.g. "AP1") the regex must allow for optional characters before or after this substring (like so ".*AP1.*"). (Regex matching is case-insensitive.) The "is in" and "matches in" operators expect the operand to be a set with multiple values, for instance a Text Variable with multiple lines (each line will be treated as a separate value), a Collection or a Text Map (which possibly contains multiple comma-separated values for each entry). NB: If a region does not have a defined value for the property, the following default values will be used: 0 (numeric properties), an empty string (text properties) or FALSE (boolean properties). Conditions based on comparison with other regions
The distance between two regions is the number of bases between them, so two regions that lie back to back have a distance of zero. Overlapping regions have a defined distance of -1.
For the distance operators the value of Operand2 must be numeric and can be provided as usual with a constant number, Numeric Variable or Numeric Map.
The comparator (•) can be any one of the standard numeric comparison operators: =, <, <=, >, >= or "in N to M" as described above.
Although compound conditions allow multiple conditions to be linked with AND/OR, these conditions are treated completely independent of each other, and it is not currently possible to make crossreferences between conditions. Hence, it is not possible to formulate general conditions on the form "where (this region overlaps RegionTrack2) AND (the overlapping region from RegionTrack2 satisfies some other condition)". However, it is possible to make use of a few predefined qualifiers to place further constraints on the regions in RegionTrack2. The following two qualifiers can be placed between the operator and the track name for all operators except "present in" and "similar in".
The special keyword "interaction partner" can be used instead of the name of a second region track for the distance operators.
Condition based on values from a numeric track within the region
The weighted sum and average only works for motif tracks where each region is associated with a Motif. In these cases the values from the numeric track will be weighted by the information content of the corresponding position in the motif.
Examples:
Compound Conditions
It is possible to define multiple feature conditions for an operation by connecting two or more individual conditions into compound conditions using the boolean operators AND and OR.
When conditions are connected with AND, the full compound condition is only satisfied when all of the individual conditions are satisfied.
With the OR operator, the full compound condition is satisfied if at least one of the individual conditions are satisfied.
When more than two conditions are connected using both operators, the AND operator takes precedence over OR. Hence, in the example below, the full condition is satisfied if either condition 1, or condition 4, or both conditions 2 and 3 are satisfied.
Parentheses can be used to group conditions together and indicate alternative orders of operations. Conditions can be nested to arbitrary levels.
Although it is possible to negate the truth value of individual conditions with the not operator, it is not (currently) possible to negate compound conditions directly. A condition on the form "where not X and Y" will be read as "where (not X) and Y" rather than "where not (X and Y)" and parentheses can not be used to achieve the latter. To negate a compound condition, the condition must instead be rephrased as two or more separately negated conditions following De Morgan's laws.
Defining compound conditions in the GUI To add more than one condition to an operation, press the small plus-button at the far right side of the Operation dialog. 
The regular view will then be replaced with a larger box where all the conditions are organized in a tree structure. All the conditions belonging to a group are listed below the operator that connects them. The image below shows one (top-level) group of conditions connected by AND. 
By default, conditions will be combined with AND, but you can change the operator by right-clicking on it and selecting "Change Operator to:" from the context-menu. To add a new condition to a group, right-click on an operator in the tree and select "Add New Condition to Group". If you choose "Add to New Group", the current group will be nested beneath a new parent group where the conditions are connected by the chosen boolean operator. 
To edit an individual condition or remove a condition from a group, right-click on the condition itself and choose "edit" or "remove" respectively from the context-menu. If you choose "Add to New Group", the selected condition will instead be added to a nested subgroup connected by the chosen boolean operator. 
Groups of conditions are shown below (and indented with respect to) the operator that connects them. In the example below, the conditions at the top level are connected by AND. This includes the first condition and a nested group connected by OR (bottom two conditions). This tree structure thus corresponds to the condition "where (region inside RepeatMasker AND (region's average Conservation < 0.5 OR region's type is in Upregulated))". You can expand or collapse nested subgroups if you like by clicking on the arrow before the operators. 
Selection Windows
Selection windows can be used to limit operations on feature datasets to within manually defined sequence segments.
These segments will usually be selected in the GUI using the selection tool.
Whenever selection windows are defined on one or more sequences, the operation dialog may contain an additional checkbox that allows
the operation to be limited to these selection windows. The selection windows themselves are listed in the textbox behind the checkbox.
Each selection window is defined on the form "sequencename:start-end" (where start and end are genomic coordinates).

Within protocols, selection window conditions are introduced by the keyword "within" followed by the list of selection windows enclosed in brackets. If other types of conditions are also applied to the same operation, the selection windows condition should always be last.
Subset Conditions
Subset conditions can be used to limit operations on feature datasets to a subset of the sequences or to limit transformations of maps
to a subset of the entries. The subset must be defined as a collection (of the applicable type) which can be then be selected from a drop-down menu in operation dialogs.

Within protocols, subset conditions are introduced by the keyword "in collection" followed by the name of the collection. If other types of conditions are also applied to the same operation, the subset condition should be listed after feature conditions but before selection windows.
Individual Operationsanalyze
The "analyze" operation performs a chosen analysis and returns an Analysis object containing the
results. The Analysis object can be inspected in MotifLab's graphical user
interface or it can be output to a text document, either in HTML-format
(possibly containing graphs and other images) or in a "raw" format which
will be suitable for parsing by other programs. In MotifLab v2, analyses can
also be output in Excel format. Note that for some analyses, the output
may change depending on the current visualization settings. For instance,
the Benchmark analysis (and several others) will use the foreground colors
of tracks when drawing charts, and hidden tracks will not be included in
the output.
Information from multiple analyses that operate on the same type of
data objects may sometimes be combined into a single analysis with
the help of the collate operation.
Arguments
Examples:
See also: benchmark, compare clusters to collection, compare collections, compare motif occurrences, compare motif track to numeric track, compare region datasets, compare region occurrences, count module occurrences, count motif occurrences, count region occurrences, evaluate prior, GC-content, motif collection statistics, motif position distribution, motif regression, motif similarity, numeric dataset distribution, numeric map correlation, numeric map distribution, region dataset coverage, single motif regression, collate apply
The "apply" operation will apply a sliding window function to a Numeric Dataset to smooth the track or to find peaks, valleys or edges in the data.
The operation goes through each position in the track in turn and defines a "window" region around each target position.
The selected window function dictates how a new numeric value can be calculated based on the values of the positions within the current window,
and the resulting value is assigned to the target position.
Arguments
Conditions: position condition Examples:
collate
The collate operation can be used to combine information from several
different analyses (or Maps) by extracting columns of data from each
analysis and putting them together in a larger table. A collated analysis
is based around a fundamental data type (Motif, Module or Sequence) and
contains rows for each of the data objects of that fundamental type. Only
information from analyses and maps that have compatible fundamental types can be collated,
and additional properties from the fundamental data objects themselves can
also be included in the final table.
Arguments
Examples:
See also: analyze combine_numeric
Combines multiple Numeric Datasets into a single track, multiple numeric maps into one map or multiple variables into one variable.
The value assigned to the target data object could either be based on the minimum value across all source data objects (inputs), the maximum value, the average value, the sum of values or the product of values.
If the source objects are Numeric Datasets the tracks are combined position by position, i.e. the value of each position in the resulting target track will be either the minimum, maximum, average, sum or product of the values
in that position across all the source datasets. If a condition is specified, only positions that satisfy the condition are combined, and positions that do not satisfy the condition are assigned the value of the first source dataset in the position.
If the source objects are Numeric Maps, these will be combined across entries for the same key. E.g. for a Motif Numeric Map, the value in the target map for motif "M00001" will be based on the values for this motif in the source maps.
The default values from each map will always be combined in the same way as the individual entries.
Arguments
Conditions: position condition Examples:
See also: combine_region combine_regions
Combines regions from multiple Region Datasets into a single track. Each sequence in the resulting track will contain the union of regions
found in all the source datasets for that sequence.
Conditions: region condition Examples:
See also: combine_numeric, merge convert
This operation can be used to convert a Numeric Dataset into a Region Dataset or vice versa.
When converting a Numeric Dataset into a Region Dataset, the regions will be based on stretches of the sequence
that satisfy a given condition. Hence, if no conditions are specified the resulting track will contain no regions.
The most natural way to convert a numeric track into regions would probably be to create regions based on stretches of the numeric track
that have values greater than zero, so this condition will be set up by default in the operation dialog. The score of each region can be specified as an argument,
and this can either be a constant value (the same for each region) or the score can be based on the minimum, maximum, average, median or sum of the values of
a numeric track within the region (the track used for this score would naturally, but not necessarily, be the same as the source track).
When converting a Region Dataset into a Numeric Dataset, positions that are not within any regions will be assigned the value 0, and positions
that are within regions can be assigned a chosen value which can be either a constant value, the value from a selected numeric track at that position,
the number of regions in the source track overlapping with that position, the highest score among all regions in the source track overlapping that position,
the sum of the scores of all regions in the source track overlapping that position, or the length of the longest region in the source track overlapping that position.
Arguments
Conditions: position condition or region condition Examples:
See also: count copy
The "copy" operation can be used to create an identical copy of an existing data object.
Examples:
count
The "count" operation counts the number of regions that overlap with a sliding window along the sequence and
returns a new numeric track containing the result for each position. For
each position in the sequence, the operation places a window of chosen size around
that position and finds all the regions that either overlap or lie fully
within this window. A value is calculated from these regions, either based
on just a count of the number regions or by summing up the scores
for all of these regions, and the resulting value is assigned to the position.
Arguments
Conditions: position condition Examples:
crop_sequences
This operation (introduced in MotifLab v2.0) will either crop the ends of the current
sequences by a specified number of bases in one or both directions, or crop the
sequences so that they align with the edges of the first and last regions
of a specified region track.
It works similarly to the "Crop Sequences" tool, but unlike that tool this
operation can also be applied to a subset of the sequences.
Arguments
Examples:
decrease
The "decrease" operation is a subtraction operator which will decrease the
value (or values) of a numeric data object by a specified amount. For
Numeric Datasets the operation will be applied to all positions in the
sequences, and for Numeric Maps it will be applied to all entries in the
map (including the "default" value).
The operation can also be applied to Region Datasets to decrease the value
of numeric properties of regions or to remove strings from a "text"
property. By default the operation will be applied to the
"score" property of the regions unless a different property is specified.
Arguments
Conditions: position condition or region condition Examples:
See also: increase, multiply, divide, set delete
The "delete" operation can be used to delete data objects that are no longer needed.
Its primary use is within protocols scripts to free up memory resources.
The operation can be applied to multiple target objects at once.
Examples:
difference
The "difference" operation will compare one data object to another object
of the same type and return a new data object highlighting the differences between the two objects.
Arguments
Examples:
See also: compare collections, compare region datasets, numeric map correlation, compare region datasets, motif similarity, benchmark, decrease discriminate
The "discriminate" operation takes a regular positional priors track as
input and turns it into a "discriminative prior" track which takes into
account the priors value of potential motifs in a set of positive
sequences (expected to contain binding sites for the target TF) compared
with a set of negative sequences (not expected to contain binding sites for this TF). For given k-mer
sequence, the discriminative prior score is defined as the ratio between the
sum of the priors scores for all the occurrences
of this k-mer in the positive set versus the sum of the prior score for the
occurrences of the same k-mer in both the positive and negative sets.
See the following references for more information: Narlikar L, Gordân R and Hartemink AJ (2007) "A Nucleosome-Guided Map of Transcription Factor Binding Sites in Yeast", PLoS Computational Biology 3(11):e215 Gordân R and Hartemink A (2008) "Using DNA duplex stability information for transcription factor binding site discovery", Pacific Symposium on Biocomputing 2008:453-464
Arguments
Conditions: position condition Examples:
distance
The "distance" operation will return a new Numeric Dataset where the value at each position in the track is determined by its distance from a selected anchor point.
The anchor point can be a fixed (or relative) coordinate position, a property of the sequence (such as the upstream or downstream end of the sequence or the TSS of the associated gene),
or the anchor point can be the nearest region in a selected Region Dataset.
Arguments
Examples:
divide
The "divide" operation is a division operator which will divide the
value (or values) of a numeric data object by a specified amount. For
Numeric Datasets the operation will be applied to all positions in the
sequences, and for Numeric Maps it will be applied to all entries in the
map (including the "default" value).
The operation can also be applied to Region Datasets to divide the value
of numeric properties of regions or to remove strings from a "text"
property. By default the operation will be applied to the
"score" property of the regions unless a different property is specified.
Note that if the "amount" argument (divisor) has a value of 0 for an entry, the division will not be carried out but the original value will be retained for that entry.
Arguments
Conditions: position condition or region condition Examples:
See also: increase, decrease, multiply, set drop_sequences
This operation can be used to completely delete a set of sequences that are no longer needed in subsequent analyses.
The operation will delete the specified Sequence Collection and all the Sequences within that collection.
Also, any other data or references related to these sequences in other Collections, Partitions, Maps or Feature Datasets will also be deleted.
Examples:
ensemblePrediction
The "ensemblePrediction" operation takes motif/binding site predictions generated by several different motif discovery programs
as input and return "consensus motifs".
The operation will return both a Motif Collection containing the consensus motifs
as well as a Region Dataset containing the locations of these motifs in the sequences (binding sites).
The actual motif prediction will be performed by an external program, and users can select which ensemble prediction method
they like to use from a list of installed programs. To configure additional ensemble prediction methods, go to the "Configure"
menu in MotifLab and select "External Programs...".
Arguments
Examples:
See also: motifScanning, moduleDiscovery, moduleScanning, ensemblePrediction execute
The "execute" operation allows MotifLab to run an external data
processing program. MotifLab can pass on any data that the program
requires and create new data objects based on the results output by the
program. This operation can thus extend the data
processing capabilities of MotifLab beyond the operations already
provided. In order to run a program with this operation, the interface of the
program must be described in XML-formatted configuration files. Ready-made
configration files for some programs are already available from the
MotifLab web site (under "Tools") or in the "External programs repository"
found under "External Programs" in MotifLab's "Configure" menu.
Examples:
See also: motifDiscovery, motifScanning, moduleDiscovery, moduleScanning, ensemblePrediction extend
Extends the size of regions in a Region Dataset in one or both directions. The regions can be extended by a fixed number of bases,
or they can be extended (one base at a time) as long as a given condition is satisfied. Note the regions will never be extended past the edge of the associated sequence.
Arguments
Conditions: position condition or region condition Examples:
extend_sequences
This operation (introduced in MotifLab v2.0) extends the current
sequences by a number of bases in one or both directions. It works
similarly to the "Extend Sequences" tool, but unlike that tool the
operation can also be applied to a subset of the sequences. Note
that the extend_sequences operation can not be used if the sequences have
associated feature annotation tracks (since MotifLab will not fill in the
missing data).
Arguments
Examples:
extract
The "extract" operation will extract a value or property (and sometimes
also new derived values) from an existing data object and return this information as a new data object.
The value or property to be extracted must be registered as an "exported property" in the source object, and different types of data objects
will export different properties. For example, it is possible to extract
the value of a single entry in a Numeric Map as a Numeric Variable,
or extract the "top X" entries
in the map as a collection. Analysis objects often export results as
Numeric Maps and Numeric Variables.
Arguments
Examples:
See also: Data filter
Removes regions that satisfy a given condition from a Region Dataset. If no condition is specified, all the regions in the dataset will be removed.
Conditions: region condition Examples:
increase
The "increase" operation is an addition operator which will increase the
value (or values) of a numeric data object by a specified amount. For
Numeric Datasets the operation will be applied to all positions in the
sequences, and for Numeric Maps it will be applied to all entries in the
map (including the "default" value).
The operation can also be applied to Region Datasets to increase the value
of numeric properties of regions or to append new strings to a "text"
property. By default the operation will be applied to the
"score" property of the regions unless a different property is specified.
Arguments
Conditions: position condition or region condition Examples:
See also: decrease, multiply, divide, set interpolate
The "interpolate" operation can be used to fill in "missing values" in a
Numeric Dataset that only contains (non-zero) values for a few discrete
positions. For example, if the values in the track are based on a
tiling-array experiment that only returns one value for each consecutive X bp region
in the sequence and only the first position in each region is assigned the value whereas
the next X-1 positions are set to 0, values for the remaining positions
can be filled in by interpolation. The default behaviour of the operation
is to interpolate between discrete, consecutive non-zero positions in the sequence
(which assumes that no position should be zero). However, it is also
possible to specify a maximum distance between the non-zero positions,
so that interpolation will not be performed when the distance
between two consecutive non-zero positions exceed this limit.
If the distance between the discrete positions that are supposed to have
legitimate values is fixed and known, it is
possible to specify this as a parameter. The operation will then locate
the first non-zero position in the sequence and assume that the next
positions to interpolate between occur periodically after this position.
This means that zero-valued positions will also be allowed.
Arguments
Conditions: position condition Examples:
See also: apply mask
Masks bases in a DNA sequence by replacing the letters in the sequence with either upper- or lowercase versions of the original letter, a new specified letter or random bases sampled from a background model.
Arguments
Conditions: position condition Examples:
See also: Background Model merge
Merges regions within each sequence that are located closer than a specified distance apart from each other.
The operation can merge overlapping regions, but also regions that are separated by gaps (in which case the resulting region
will cover the full span of the merged regions, including the gaps).
If the merged regions have the same type, the resulting region will also have this type, else the region is assigned the type "merged".
If the merged regions have the same orientation, the resulting region will also have this orientation, else the region is assigned the orientation "undetermined".
The score or the resulting region will be assigned the score of the highest scoring region among those merged.
Arguments
Conditions: region condition Examples:
See also: combine_regions moduleDiscovery
The "moduleDiscovery" operation can be used to perform 'de novo' module discovery in a set of sequences,
meaning that it can search for possible modules (combinations of binding motifs) that are present in the sequences
without having prior knowledge about what the modules look like.
The operation will return both a Module Collection containing the discovered modules
as well as a Region Dataset containing the locations of these modules in the sequences.
The actual module discovery will be performed by an external program, and users can select which module discovery method
they like to use from a list of installed programs. To configure additional module discovery methods, go to the "Configure"
menu in MotifLab and select "External Programs...". Note that this operation can be applied to both DNA Sequence Datasets
and Region Datasets, but which type of source data to use will depend on the chosen module discovery method.
Arguments
Examples:
See also: moduleScanning, motifDiscovery, motifScanning, ensemblePrediction moduleScanning
The "moduleScanning" operation can be used to search DNA sequences for matches to a set of predefined modules.
The operation will return a Region Dataset containing the locations of these modules in the sequences.
The actual module scanning will be performed by an external program, and users can select which module scanning method
they like to use from a list of installed programs. To configure additional module scanning methods, go to the "Configure"
menu in MotifLab and select "External Programs...". Note that this operation can be applied to both DNA Sequence Datasets
and Region Datasets, but which type of source data to use will depend on the chosen module scanning method.
Arguments
Examples:
See also: moduleDiscovery, motifScanning, motifDiscovery, ensemblePrediction motifDiscovery
The "motifDiscovery" operation can be used to perform 'de novo' motif discovery in a set of sequences,
meaning that it can search for possible binding motifs that are present in all or several of the sequences
without having prior knowledge about what the motifs looks like.
The operation will return both a Motif Collection containing the discovered motifs
as well as a Region Dataset containing the locations of these motifs in the sequences (binding sites).
The actual motif discovery will be performed by an external program, and users can select which motif discovery method
they like to use from a list of installed programs. To configure additional motif discovery methods, go to the "Configure"
menu in MotifLab and select "External Programs...".
Arguments
Examples:
See also: motifScanning, moduleDiscovery, moduleScanning, ensemblePrediction motifScanning
The "motifScanning" operation can be used to search DNA sequences for matches to a set of known motifs.
The operation will return a Region Dataset containing the locations of these motifs in the sequences (binding sites).
The actual motif scanning will be performed by an external program, and users can select which motif scanning method
they like to use from a list of installed programs. To configure additional motif scanning methods, go to the "Configure"
menu in MotifLab and select "External Programs...".
Arguments
Examples:
See also: search, motifDiscovery, moduleDiscovery, moduleScanning, ensemblePrediction multiply
The "multiply" operation is a multiplication operator which will multiply the
value (or values) of a numeric data object by a specified amount. For
Numeric Datasets the operation will be applied to all positions in the
sequences, and for Numeric Maps it will be applied to all entries in the
map (including the "default" value).
The operation can also be applied to Region Datasets to multiply the value
of numeric properties of regions or to append new strings to a "text" property.
By default the operation will be applied to the
"score" property of the regions unless a different property is specified.
Arguments
Conditions: position condition or region condition Examples:
See also: increase, decrease, divide, set new
Creates a new data object according to given specifications.
Exactly how to define an object depends on the type, and most types of objects can be constructed in several different ways.
For instance, a background model can be created by explicitly specifying its order and all the oligo-frequencies, or it can be generated automatically based on a DNA sequence.
Two modes of creation are supported by all types of object, namely creating an object based on a file (in an applicable data format) and creating an object based on a Text Variable or Output Data object containing a similarly formatted file. The syntax for these modes are almost identical:
NewObject = new <data type> (File:"path/to/file", format=<formatname> {format arguments} )
NewObject = new <data type> (Input:DataObjectName, format=<formatname> {format arguments} )
The data object referred to by the "Input:" mode must be either a Text Variable or Output Data object.
The format arguments (enclosed in braces) are optional, and default values for that format will be used if left out.
The format name is also optional, and the default format for the datatype will be used if left out (with default format arguments).
Arguments
Examples:
See also: Text Variable, Output Data normalize
The normalize operation rescales the numeric values of a data
object from one range to another. The operation can currently only be
applied to Numeric Datasets and Region Datasets (for the latter it will be
applied to the score-property), but it will be updated in the future so that it
will also work on Numeric Maps. It has two different modes of normalization:
"normalize sum to one" or "normalize to range". The first mode will scale all the
values so that the sum total equals 1.0 (and the values thus form a probability
distribution), while the second mode will scale the values from one range
("old range") to another ("new range").
Arguments
Conditions: position condition or region condition Examples:
output
Outputs data items to text documents in selected data formats. The document will be wrapped in a so-called "Output Data" object and the
contents of this can be saved to files. If MotifLab is run in CLI-mode (without the GUI), all Output Data objects that are created during the
execution of a protocol script will automatically be saved to files after completion of the protocol (the filename will be the name of the data object with a suffix determined by the data format used).
If no target Output data object is specified for the output operation, a new Output object will be created automatically and assigned a default name consisting of the prefix "Output" followed by an incremental number.
If a target Output data object is specified and it already exists, the output will be appended to that object if possible. If it is not possible to append more text to this data object (because it is formatted in a data format that does not
allow additional text to be appended, such as HTML-formats), the operation will end with an error.
When Feature Datasets are output, the sequences will be output in the order they are currently sorted.
Graphics output Output saved in Excel and HTML formats may include graphics, such as charts and motif logos. In Excel, these graphics will always be embedded in the file itself, but in HTML format you have a few options on how to output motif and module logos. Please refer to the HTML format documentation for more information. Direct output Version 2.0+ of MotifLab allows the output operation to be used to output literal text strings directly to output objects (but only within protocols). The format is: <Output object> = output "some text string...". The text string enclosed in double quotes can contain references to data objects on the form "{dataobject} and the value of the referenced object will then be included in the output as explained in the documentation for the Template and TemplateHTML data formats (the same formatting-options for referenced objects are also available). The text string can also contain TABs, newlines, double quotes and backslashes if these are properly escaped as \t, \n, \" and \\ respectively.
Arguments
Examples:
See also: Output Data, FASTA, GFF, EvidenceGFF, BED, WIG physical
The "physical" operation estimates different physical properties of the
DNA double helix based on local sequence composition and returns a Numeric
Dataset containing a value for the selected property for each position.
For each position in the sequence, the value of the physical property is
estimated by examining the nucleotide composition within a window
region around that position. Depending on the selected property, the resulting value
is either derived directly from the base (or oligo) frequencies or it is
estimated by summing up values based on a smaller sliding window (2 or 3
bases long) within the larger window region.
Arguments
Conditions: position condition Examples:
plant
The "plant" operation can be used to create artificial
benchmark datasets with known TFBS regions to test the performance of motif
or module discovery methods. The operation will take a DNA sequence (which
can be real or artificial) as input, insert new motif sites at random
locations in the sequence and return the updated DNA sequence along with a Region Dataset containing the planted
sites. Either a single motif or module or a collection of up to five
different (non-overlapping) motifs can be planted in each sequence according
to specifications.
Arguments
Examples:
predict
The predict operation can make use of trained Priors Generator objects to derive "positional priors" tracks where the value
of each position in the track can be interpreted as a prior probability of observing a specific feature at that position.
The feature which is predicted is already set in the Priors Generator and all tracks that the Priors Generator needs
in order to predict the target feature must also be available in order to use the operation. These inputs are not explicitly
declared but must have the same name and types as the original tracks used when training the Priors Generator.
E.g. If a Priors Generator was trained to predict the locations of transcription factor binding sites,
on the basis of three tracks named respectively "Conservation", "DNaseHS" and "ChipSeq", the same three tracks must also
be available in order to use the predict operation with this Priors Generator.
Arguments
Examples:
See also: Priors Generator prompt
The "prompt" operation can be used in protocol scripts to provide users with some control and allow them to interactively select new values for different data objects
during the execution of the protocol. When a "prompt" command is encountered in the protocol, a dialog box will appear and ask the user to select a value for the data object.
Note that the target data object must already exist (the prompt operation can not be used to create new data objects)
but the object can be "empty". The current value of the data object will be used as the default value, and this value will be displayed to the user who
can decide to keep the data object as it is or select a new value for it.
Arguments
Examples:
prune
The "prune" operation can be used to remove duplicate regions from a
Region Dataset. These duplicates can either be regions that are exactly
identical to another region in the same track or they can be overlapping
regions for motifs that are considered to be similar to each other (and
hence duplicate predictions of the same TF binding site). The operation
searches for groups of duplicate overlapping regions and removes all but
one of the regions in each group.
Arguments
Examples:
rank
The "rank" operation will return a new Numeric Map where the values correspond to the rank order of the entries in another Numeric Map,
a similar numeric column from an Analysis, or internal numeric properties of data objects. The rank order can also be based on a weighted
combination of several such properties. In that case, each property is first ranked on its own and the rank-values are multiplied by the weight
for that property (if specified). The ranks are then summed up across all properties and a final rank order is derived from these values (in ascending order).
(Note that the entries are not ranked first by the first value, and then by the second value to break ties etc.)
Entries that have the same value will receive the same rank. For example, a map with entries "A=3,B=5,C=2,D=13" will be ranked (ascending) as "A=2,B=3,C=1,D=4",
and a map with entries "A=3,B=3,C=2,D=13" will be ranked (ascending) as "A=2,B=2,C=1,D=4" (Note that D is still ranked as number 4 and "rank 3" has been skipped).
Arguments
Examples:
replace
The "replace" operation (v2.0) replaces portions of text in a Text Variable or a textual property of a Region Dataset.
The basic mode of this operation will search the body of text for a specified search term (which can be in the form of a regular expression)
and replace all instances matching this search term with a given replacement text (which can contain backreferences to capture groups in the search expression).
The operation can also be used to search Text Variables for instances of macro names and replace these with their corresponding definitions ("replace macro") or to add new lines to the beginning or end of a Text Variable ("replace beginning/end").
Arguments
Conditions: region condition Examples:
score
The "score" operation uses a basic motif scanning algorithm to compare a
single motif model (or a collection of motifs) against a DNA sequence, but
rather than returning a track containing matching regions, the operation
returns a numeric track with the motif match score for each position.
If the operation is used with a collection of motifs rather than a single
motif, all the motifs in the collection will be scanned against the DNA
sequence and the highest match score obtained for each position will be returned.
Arguments
Conditions: position condition Examples:
See also: motifScanning search
This operation can be used to search DNA sequences for occurrences of a given DNA sequence pattern (or multiple patterns),
specified as either regular expressions (in JAVA syntax) or as IUPAC consensus patterns.
The search pattern can be a literal string enclosed in double quotes or the name of a Text Variable, single Motif or a Motif Collection (without quotes).
When searching for a Motif or Motif Collection, the operation will search for the "consensus sequence" representation of the Motif (or all the motifs in the collection).
The operation can also be used to search for occurrences of tandem or inverted repeats (two identical DNA patterns that occur close to each other in the DNA sequence).
Constraints can be placed on the size of the two halfsites and the size of the gap between them.
Arguments
Examples:
See also: motifScanning set
The "set" operation is an assignment operator which can be used to set the
value of a numeric data object to a new specified value. For
Numeric Datasets the operation will be applied to all positions in the
sequences, and for Numeric Maps it will be applied to all entries in the
map (including the "default" value).
The operation can also be applied to Region Datasets to set the value
of numeric properties or text properties. By default the operation will be applied to the
"score" property of the regions unless a different property is specified.
Arguments
Conditions: position condition or region condition Examples:
See also: increase, decrease, multiply, divide split_sequences
This operation (introduced in MotifLab v2) can take an existing set of sequences and derive a new set of sequences based on subsegments of the originals. The original sequences can be kept together with the new sequences or optionally be deleted.
The subsegments on which to base the new sequences are taken from the locations of regions in a specified region track. Each region in this track will give rise to one new sequence, so if two regions are overlapping they will result in two overlapping sequences.
The new sequences will have names on the form "XXX_n" where XXX is the name of the original sequence and n is an incremental number starting at 1 for each original sequence.
Note that the new sequences are not allowed to extend beyond the edges of the original sequences even if the regions they are based on do that. For example, if you have a sequence spanning the [-1000,+200] region around the TSS of a gene which is 2000bp long (thus extending 1800bp further downstream of the original sequence) and you use split_sequences to create new sequences based on the gene annotation track, the new sequence location will be the intersection of the old sequence and the gene region, meaning the new sequence will correspond to the 200bp region starting at the gene TSS and extending downstream to the end of the original sequence. The gene region is kept at its original length, however, and is allowed to extend past the edge also in the new sequence. The operation will return a sequence partition object where each newly created sequence is assigned to a cluster named after the original sequence it was based on. Old sequences not created by split_sequences will not be assigned to any cluster in the partition. The result of applying split_sequences is usually some form of cropping of the original sequences (and also all associated feature tracks) so in some ways it is similar to the crop_sequences operation. The difference between this operation and crop_sequences is that the latter only modifies the original sequences whereas split_sequences creates new sequences. If your sequences contain exactly one region each, the result of the two operations will be (almost) the same. However, if you have a sequence containing two regions, crop_sequences will crop the original sequence so that it begins at the start of the first region and ends at the end of the second region, whereas split_sequences will create two new sequences where each is cropped to match one of the regions in the original sequence. The split_sequences operation is the only exception to the rule that new sequences cannot be created after feature datasets have been added. The reason for having this rule is that the feature tracks would normally be undefined within the new sequences. However, since the sequences created by split_sequences are based on subsegments of existing sequences, all the necessary feature data for the new sequences will already be present.
Arguments
Examples:
statistic
Calculates a statistic for each sequence in a dataset and returns a Sequence Numeric Map containing the results for each sequence.
Arguments
Conditions: position condition, region condition or subset condition Examples:
threshold
Assigns all numeric values in a data object that are equal to or above a
specified cutoff threshold a new value and those below the cutoff a
different value. For Numeric Datasets the operation will be applied to
every position in all sequences, for Region Datasets the operation will be
applied to the score-property of every region, and for Numeric Maps and
Expression Profiles the operation will be applied to every value in the Map/Profile.
Arguments
Conditions: position condition or region condition Examples:
transform
Transforms each numeric value in a data object according to a selected
mathematical function. For Region Datasets the transform will be applied
to the 'score' properties of the regions unless a different numeric property is specified.
A few special transforms that target Region Datasets may also modify non-numeric values ("reverse" and "type-replace").
Note that values which can not be transformed for some reason will just be skipped (e.g. when taking the logarithm of negative values or dividing by zero).
Usually, a warning message will be provided in the log when this occurs.
Arguments
Conditions: position condition or region condition Examples:
See also: distance ProtocolsA "protocol" is a document which describes a list of operations to be executed in order (including specifications of their parameters, conditions and constraints). Protocols can be used to document the steps you perform during an analysis session, and they can describe workflows that can be automatically executed by MotifLab. If you like, you can specify exactly which sequences to perform the analyses on in the protocol itself, and the protocol will then always perform the analysis on these squences. However, if the sequences are not explicitly specified, the protocol will just describe a generic analysis workflow which can be applied to any set of sequences (as long as any additional data needed by the protocol is available for the organism and genome build you apply the analysis to).Creating a protocolProtocols can either be written manually in the protocol editor (or an external text editor) or they can be made with MotifLab's record functionality which will automatically register all the operations you perform to the protocol.To create a new protocol, press the "New Protocol" button in the toolbar or go to the "File" menu and select "New Protocol" from there. The protocol editor (described below) will then display the new protocol. You can also open a previously saved protocol by pressing the "Open Protocol" button in the toolbar or selecting "Open Protocol" under the "File" menu. To activate the "record mode", simply press the round red record button in the toolbar (or select "Record" under the "Protocol" menu). Any operations you perform after activating record mode will be registered in the protocol. Note that the recorded protocol commands will be inserted at the location of the cursor in the editor and not appended to the end (unless the cursor is at the end of the protocol). This means that you can also use record mode to insert new commands anywhere in the protocol by first placing the cursor at a line and then performing a new operation. Press the stop button in the toolbar to deactivate record mode (or select "Stop" under the "Protocol" menu). Executing a protocolYou can execute a protocol by pressing the "Execute" (play) button in the toolbar or selecting "Execute" from the "Protocol" menu. MotifLab will then go through all the operations that are described in the protocol. If the protocol contains operations that applies to sequences and no sequences are defined in the protocol itself, the protocol will be applied to the sequences that are currently known to MotifLab. If no sequences are known, MotifLab will display the Sequence Dialog and prompt the user to specify which sequences to perform the protocol on.It is also possible to execute just a subset of the commands listed in the protocol. To execute a number of consecutive lines, select the lines that you want to run by marking the text in the protocol editor (you need not select the full line to include it, it is enough that just parts of a line is selected). Then go to the "Protocol" menu and select "Execute Current Selection". You can also execute only the line where the cursor is currently at by selecting "Execute Current Line" from the "Protocol" menu (NB: this might not work properly in version 1.000 due to a bug), or by holding down the CONTROL key while pressing ENTER inside the protocol editor (if you hold down the SHIFT key at the same time you will suppress any dialogs that might pop up to display the results of the operation).To stop the execution of a protocol before it is finished, just press the "Stop" button in the toolbar. The protocol languageThe standard protocol language employed by MotifLab was designed to be close to natural language so that it should be possible for a human user to read and understand a protocol script without being an experienced programmer. However, the protocol language also has a few constraints in order to make it easily processable by MotifLab. First, each line in the protocol can only contain one command and each command can not span more than one line. Second, the first word of a command (after the assignment operator "=") must be the name of an operation. Apart from that, each operation decides for itself how the command should be expressed. However, most operations rely on a command syntax which follows this general format:
The target clause at the start of the line states a name for a new data object that is created by the operation. For many operations this target clause is optional and the target will then be the same as the source object. E.g. in the first example command line below, the value of X is increased by 10, since X is both the source object and the (implicit) target. The command in the second line, however, will create a new data object named Y which has a value equal to X+10, but the value of X itself will not be changed. Some operations return more than one data object and in such cases the target is specified as a vector with multiple comma-separated names enclosed in square brackets (as can be seen for the "plant" operation in the last example below). The arguments clause specify values for different arguments used by the operation. This will almost always include the "source" data object that the operation should be applied to, but different operations may also require additional arguments to be specified. For example, when performing the "increase" operation on a Region Dataset, the operation also requires two additional arguments to be specified: one which tells the operation which property of the regions to increase the values of and another which tells the operation how much the current value of this property should be increased. Some operations have rather many arguments (or even a variable number of arguments) and these operations often rely on "argument maps" to specify values for some or all of their arguments in a more compact form. An argument map is simply a comma-separated list of "argumentName=argumentValue" pairs enclosed in curly braces. The two last example commands below make use of argument maps (operations "motifScanning" and "plant"). The condition clause is always optional but can be used to limit the application of the operation. Depending on the type of condition, this clause will either begin with "where" or "in collection". In the following example commands the operation name is shown in red, the arguments clause in green, the condition clause in blue and the target clause in pink.
Comments Lines in a protocol that start with a # sign will be treated as comments and ignored during execution. Note that all comments must be on their own lines since it is not possible to add comments at the end of other command lines. Temporary data objects Sometimes it will be necessary for a protocol script to create temporary data objects that are used for e.g. intermediate calculation steps but are not really interesting for the user after the execution of the protocol has ended. Such data objects can be given names starting with an underscore to mark them as temporary. Temporary data objects will not be displayed in any data panels or in the Visualization panel and they will be deleted immediately after the protocol execution ends. Flow controlProtocols scripts in MotifLab are designed to be conceptually simple, where each line in the protocol from the first to the last should be executed once and only once in succession. The protocol language and commands to perform various operations are inspired by the paradigm of declarative programming, whereby a programmer describes what they want to achieve rather than micromanaging exactly how to go about to produce the desired outcome. For example, if an operation is applied to a data object that naturally contains subentries, MotifLab will implicitly perform the operation on each of these subentries in turn, as long as all imposed conditions hold true for the entry. Because of this (and also because MotifLab does not support constructs such as data arrays or reference variables), there is really little need for the protocol language to include flow-control statements such as loops and conditional blocks.Nevertheless, from version 2.0 onwards, MotifLab does support limited flow-control in the form of conditional "if-then-else" statements. The basic syntax for a conditional statement block is:
You can have alternative "else if" condition blocks after the first "if", and the first block whose condition is satisfied will then be executed. An optional default "else" block will only be executed if none of the conditions for any of the previous "if" or "else if" blocks were satisfied. It is allowed to nest "if-else" statements to arbitrary levels.
So far it is only possible in the condition expression to compare a single data object to another data object or literal value (textual or numeric). However, multiple conditions can be connected with boolean operators "AND" and "OR" to create more complex compound conditions. Conditions allowed in flow-control statements
When Operand2 is "Text" the operand can either be a Text Variable, a Text Map (in which case only the default value is considered), a Collection or a literal text enclosed in double quotes. When the "equals" comparator is used, the two bodies of text must represent identical documents, but for the other comparators the bodies of texts are considered as "sets of strings" and the order of the strings is not important. For example, if T1 is "apples,oranges" and T2 is "oranges,apples" then "T1 equals T2" is false but "T1 = T2" is true. When Operand2 is "Numeric" the operand can either be a Numeric Variable, a Numeric Map (in which case only the default value is considered), or a literal number. For data objects that are not Text Variables, Numeric Variables, or Collections, only the two comparators "=" and "<>" are available to determine if the objects represent the same value or not (the definition of representing the "same" value depends on the data type). Example: When the protocol below is executed, the user will be asked interactively for which motif scanner to use to predict binding sites (via the prompt command). Depending on the choice of algorithm, which can be either "MATCH" or "SimpleScanner", only one of the two motifScanning commands will be performed and return a BindingSites track.
The protocol editorThe protocol editor can be found under the "Protocol" tab in the main panel.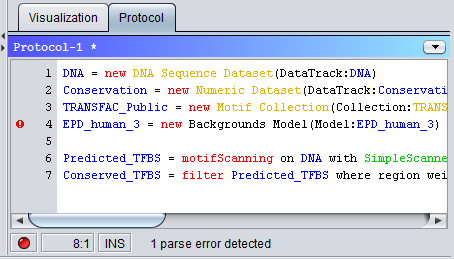 The protocol editor consists of three panels. On the top is a blue header panel which displays the name of the protocol. New protocols are given default names like "Protocol-1", "Protocol-2" etc., but you can change the name by saving the protocol to a file (by going to the "File" menu and selecting "Save" or "Save As..."). The protocol will then be given the same name as the file that you saved it to (minus the file-suffix). A protocol which has not been saved yet (or has been changed since it was last saved) will have an asterisk after the protocol name in the header. It is possible to have multiple protocols open at the same time in MotifLab, and you can then switch between them via a drop-down menu which is available by pressing the down-arrow button on the right side of the header (or by going to the "Protocol" menu and selecting "Change Protocol"). Only the protocol which is currently displayed in the protocol editor will be "active", however. The main part of the protocol editor is the editor panel itself. Here the currently selected protocol is displayed and can be edited. Each operation command must be written out on a single line in the protocol in order for MotifLab to understand it correctly. (Word wrapping functionality for long lines will hopefully be included in a future version of MotifLab). The protocol editor can use colors to highlight keywords of different types in the protocol. According to the default color scheme, the names of operations are colored RED, names of specific data objects are colored BLUE, general data types are in ORANGE (as are names of analyses and names of general data formats for input and output), names of external programs are in GREEN, literal numeric constants are in PINK and literal text constants (in double quotes) are in GREEN, display settings are in CYAN and comments are in GRAY. If you don't like these default colors you can change them by selecting "Options..." from the "Configure" menu and go to the "Protocol Editor" tab in the Options-dialog which pops up. The editor panel has a gray margin area to the left which displays line numbers in front of each protocol line and sometimes also small icons in front of these line numbers. These icons have the following interpretations:
At the bottom of the protocol editor is the status panel with three boxes followed by a status message line. The first box contains a "status light" which can either be colored green, yellow or red (or black when there are no protocols). A green light means that the protocol does not contain any errors as far as MotifLab can tell, and it should therefore be possible to execute it. A red light means that the protocol contains errors which makes it impossible for MotifLab to parse it correctly. The number of errors detected in the protocol will be displayed int the status message line, and the lines that contain these errors should also be marked with error icons in the margin. (To see what is wrong with a line, point the mouse at the error icon to see the error message). If you try to execute a protocol containing errors, MotifLab will refuse and display an error message. If the status light has a yellow color this indicates that MotifLab has yet to determine whether the protocol contains any errors or not. This color is usually displayed if you start typing into the protocol. MotifLab will then wait until you have stopped typing before it checks the protocol for errors and then changes the light to either green or red. The second box on the status line (after the status light) displays the coordinates of the cursor in the format "line:column", and the third box shows if the editor is currently in "insert mode" (INS) or "overwrite mode" (OVR). If the editor is in "insert mode", newly typed characters will be inserted at the position of the cursor and any text that follows the cursor will pushed forward. If the editor is in "overwrite mode", however, any character currently under the cursor will be replaced by a newly typed character. You can toggle between the two modes by pressing the INSERT key on your keyboard (if you have one). Display settingsWhen MotifsLab's "record mode" is activated to log a users actions in a protocol, only the operations that the user executes are recorded. Other activities the user performs, such as for instance changing the color or height of a data track, are not recorded. However, it is possible to include such visual cues in the protocol as well, by manually entering display setting statements. A display setting statement starts with a dollar sign '$' (or alternatively an exclamation mark '!') at the beginning of the protocol line and is immediately followed by the name of the setting to be specified.The general format is:
$setting(target)=value
Note that both the setting and the value are normally case-insensitive but the target is case-sensitive. The difference between using a dollar sign in front of the statement and an exclamation mark, is that when the dollar sign is used, the system will check that the target data objects exist and have the correct type. If an exclamation mark is used instead, the system will not perform any checks but just make a record of the setting for future reference. Hence, using the exclamation signs allows you to set display settings for data objects that have not been created yet. A table describing all recognized display settings is provided below. The target argument specifies which data object(s) the setting should be applied to. A target can for instance be the name of a feature track, a sequence, a motif or a module depending on the display setting. A comma-separated list of targets can be specified instead of just a single target, and if the setting applies to sequences, motifs or modules, names of collections of such objects can also be used. Alternatively, instead of naming specific targets, a single wildcard (*) can be used to refer to all data objects of the applicable type. For settings that target "region types", a list of types can be provided or a special wildcard that target all region types found in a given Region Dataset like this "datasetname:*". Note that some settings do not have specific targets, in which case the target argument should be left blank. The allowed values for each display setting are also specified in the following table. Some settings require the value to be a specific keyword (such as for the "graphtype" setting), while others require a numeric (usually integer) value or a boolean value (which can be specified as either TRUE/YES/ON, or FALSE/NO/OFF). The special color value can be entered as either a comma-separated triplet of numeric RGB-values in the range 0 to 255 (e.g.: "255,0,0" for RED or "255,255,0" for YELLOW), as a 6 digit hexadecimal number preceded by # (e.g. "#FF0000" for RED or "#FFFF00" for YELLOW) or using one of the following color-keywords: BLACK, BLUE, CYAN, DARK BROWN, GRAY, GREEN, LIGHT BLUE, LIGHT BROWN, LIGHT GRAY, LIGHT GREEN, MAGENTA, ORANGE, PINK, RED, VIOLET, WHITE or YELLOW. MotifLab v2.0 also allows the color to be specified with a colon-separated triplet of numeric HSB-values in the range 0.0-1.0. In MotifLab v2.0, some of the fonts used (for instance to draw base letters in DNA tracks or tick labels in graphs) can also be changed. Fonts are specified as a comma-separated triplet defining the fontname, size and style. The fontname can either be one of the five logical fonts ("Serif", "SansSerif", "Monospaced", "Dialog" or "DialogInput") or the name of any font installed on the users computer. The size is an integer between 3 and 200 (recommended range between 8 and 30), and style can be chosen among the following options: "plain", "bold", "italic" or "bolditalic". E.g.: $setting("system.dnaFont")=Serif,12,bold.
Examples of display setting statements:
$visible(*)=YES # Shows all
current feature tracks in the visualization panel
$hideMotifs(*) # Hides all motifs so that their TFBS are not shown within motif tracks in the visualization panel $showMotifs(MotifCollection1,M00001,M00002,M00004) # Shows all motifs in the collection and 3 more $height(Conservation,RepeatMasker)=26 # Sets the height of these two tracks to 26 $margin()=10 # Sets vertical distance between sequences to 10 pixels $color(Conservation)=RED # Sets the color of the Conservation track to red $color(RepeatMasker)=#0000FF # Sets the color of the RepeatMasker track to blue $color(CCDS)=0,255,0 # Sets the color of the CCDS track to green $expanded(TFBS)=False # Turns off expanded mode for the TFBS track $order(DNA,CCDS,Conservation,RepeatMasker,TFBS) # Changes the order of the given tracks $moduleFillColor()=Type # Specifies that all modules should be colored according to their type $moduleOutlineColor()=BLACK # Sets outline color of all modules to black $hideRegion(RepeatMasker:*) # Hides all regions found in the RepeatMasker track $showRegion(AluSx,LTR2B) # Shows regions of the AluSx and LTR2B (repeat) types Display setting statements can also be used to perform a few other tasks in a protocol that are not necessarily connected to visualization. The following table contains a few such useful statements:
MacrosThe possibility of defining macros to use in protocols was introduced in MotifLab v2. Macros are named entities that can be referenced in protocol scrips, and right before a protocol is to be executed all occurrences of macros will be substituted with their respective definitions. This makes it possible to rewrite parts of a protocol on-the-fly.Macros can either be defined in the GUI's macro editor, which can be found by selecting "Macro Editor..." from the "Protocol" menu, or with the command-line argument "-macro <name> "<definition>"" in the CLI-client. Macros can also be defined within a protocol itself using a display setting command, like so
The difference between using an exclamation mark versus a dollar sign for the macro command is that the exclamation mark will always assign the new definition to the macro when the command is executed, but if you preceed the command with a dollar sign the macro will only be assigned the new definition if it is not already defined through other means (GUI macro editor or CLI-option). There are no restrictions on the name of a macro except that it cannot contain a closing parenthesis. However, it is advisable to keep the names simple and only use letters and underscores. Also, since every instance of the macro name anywhere in the protocol will eventually be replaced by its definition, you should make sure that the name is unique enough to not cause any off-target substitutions (for example if the macro name is a substring of some other word used in the protocol). There are two different kinds of macros in MotifLab, simple macros and list macros. Simple macro A simple macro will just replace every occurrence of the macro name in a protocol with the corresponding definition. For example, the following protocol contains a macro named VALUE with the definition "942".
This will result in the following protocol
List macro A list macro is defined by enclosing the macro definition in brackets. Inside the brackets you can list multiple comma-separated values. If a line in a protocol contains a list macro, MotifLab will expand that line into multiple lines with each line using the next value in the list as its macro definition. For example, the following simple protocol contains a list macro named INDEX with four listed values.
The second line in the protocol contains the macro name and will therefore be expanded into four repeated lines with each line using the next value in the list for the macro. The resulting protocol will thus look like this:
List macros can contain any kind of values, not just numbers, but using list macros to append incremental numeric suffixes to data objects and thus creating a kind of "array" of related data objects is a common scenario. For this reason, it is also possible to use the short-hand notation "[1:4]" as a list macro definition instead of listing all the numbers "1,2,3,4" explicitly. In this case, MotifLab will automatically create the list by iterating through all the numbers starting from the first value (before the colon) up to and including the second (after the colon). If the last value is smaller than the first, the numbers will appear in reverse order (e.g. the list "[7:3]" will expand to "7,6,5,4,3"). Since it is most common to start at the value 1 and go upwards, you can even drop the first value if you want in this case. Hence, the simple list macro "[10]" will expand into 10 elements numbered from 1 to 10. Note that it is possible to nest macros so that the definition of one macro contains the name of a second macro. Every time MotifLab expands a macro into one or more lines, it will check those lines over again for the presence of additional macro names and continue to expand macros until no more macros can be found. (For this reason you should avoid circular macros at all cost since that will cause MotifLab to hang). If a line in the protocol contains more than one macro, these will be expanded in a left-to-right order. If you have a protocol containing macros, it is possible to preview the resulting expanded protocol by selecting "Expand Macros" from the "Protocol" menu in the GUI. This will expand all recognized macros in the protocol and show the result in a new protocol file (having the same name as the original protocol but suffixed with "-[macro expanded]"). AnalysesWARNING: When you perform analyses on sequences, motifs or modules, the resulting analysis object will store the names/identifiers of these data objects but not necessarily other information about them. When you view an analysis or output it, MotifLab may dynamically add more information about the sequences/motifs/modules to the output based on their current values, but if you have modified or replaced these objects after doing the analysis, these properties may not reflect the actual values that the objects had when the analysis was performed!This usually only applies to individual sequences/motifs/modules and not to collections and partitions (which will normally be copied by the analysis). For instance, if you perform a "Count Motif Occurrences" with a Motif Collection containing motif M00143 and you later change the matrix values of this motif, the motif logo shown when viewing the results of the analysis will not reflect the actual motif that was used when performing the analysis. benchmarkThis analysis can be applied to: Region Dataset
Description
The benchmark analysis can be used to evaluate the performance of motif discovery programs by comparing tracks with predicted TF binding sites (or other predicted regions) returned by these programs against a track containing the "correct" answer (e.g. all known TFBS in the sequences). The analysis calculates several common performance metrics (statistics), including e.g. sensitivity, specificity, positive predictive value, F-measure and Matthew's correlation as described below. Some of the metrics (viz. sensitivity, PPV, PC, ASP and F-measure) can be evaluated at both a "nucleotide level" and "site level", whereas the remaining metrics are only defined at the "nucleotide level". The formulas for all metrics are based on four parameters that count the number of true positive instances (TP), false positives (FP), true negatives (TN) and false negatives (FN) respectively. At the "nucleotide level", a true positive is a nucleotide position that is correctly predicted as being part of a binding site (both the prediction track and answer track have regions that overlap with this nucleotide). A false positive is a nucleotide position that is within a region in the prediction track but not in the answer track (the nucleotide is wrongly predicted to be within a TFBS). A true negative is a nucleotide that is correctly predicted to not be within a TFBS but rather being part of the background sequence (it is outside regions in both the prediction and answer tracks). A false negative is a nucleotide that is predicted to be part of the background sequence when it is actually within a true TFBS (it is outside of regions in the prediction track but inside a region in the answer track). At the "site level", a region in the answer track that is overlapped by a region in the prediction track is counted as a true positive, a region in the answer track that is not overlapped by a predicted region is called a false negative and a predicted region that does not overlap with a region in the answer track is called a false positive (true negatives are not counted at the "site level"). The minimum amount of overlap between the answer region and predicted region that is required in order to call it a true positive can be specified as a parameter to the analysis. If the benchmark analysis is based on several sequences, the TP/FP/TN/FN parameters will be counted for each sequence and then summed up to produce a total for the whole dataset before calculating the statistics below.
All of these metrics, except CC, has a range between 0 (worst score) and 1.0 (best score). The CC metric has a range from -1.0 to 1.0, where a score of 1.0 means that the prediction and answer tracks are equal (at least in terms of overlapping regions), a score of -1.0 means that the prediction track is exactly the opposite of the answer track (all true regions were predicted as background and all true background nucleotides were predicted as being within TFBS). A score of 0 means that there is no correlation between the prediction track and the answer track (such a result would be expected if the predictions were based on random guessing). Some motif discovery methods are based on stochastic algorithms and may produce different results if run several times on the same dataset. For such methods it would be useful to report the average results (with standard deviation) across multiple runs. The benchmark analysis allows the results for multiple prediction tracks for the same method to be combined into a single average statistic. In order to do this, the "Aggregate" parameter flag must be set (see below) and the tracks must be given names in the format "methodname_number", i.e. the name of the track (which is often the name of the method) must be suffixed by an underscore followed by a number (which need not be incremental). For example, if you have run a method based on Gibbs sampling five times and the TFBS prediction tracks returned by this program are given the names "Gibbs_1", "Gibbs_2", "Gibbs_3", "Gibbs_4" and "Gibbs_5", the benchmark analysis will take the average score for each metric across these five tracks and present the results as a method called "Gibbs". Standard deviations are shown as error bars in the bar plot (in current versions of MotifLab the standard deviations are not reported as numbers). The analysis compares the answer track to all other Region Datasets known to MotifLab, but only results for Region Datasets that are currently visible in the GUI will be included when the Analysis object is examined or output to HTML or RawData formats. The order of the tracks in the output is based on their order in the Features Panel, and the colors used for the tracks in the bar chart are based on the current colors of the tracks. If MotifLab is run without the GUI in command-line mode, the visibility of the tracks can be set in the protocol with the "$show(trackname)" and "$hide(trackname)" display setting statements. The colors of the tracks can be set with "$color(trackname)=color" and the order of the tracks can be set with "$order(track1,track2,...,trackN)". These commands can also be used for aggregated tracks if the dollar sign is exhanged for an exclamation sign. E.g. to set the color for the aggregated "Gibbs" track based on the five tracks mentioned above, a command like "!color(Gibbs)=RED" could be used. It is also possible to specify the colors to use for the different performance metrics by using commands on the form "$setting("systemColor.Sensitivity")=RED". The standard colors for these metrics are defined in the startup script for MotifLab (go to the "Configure" menu and select "Edit Startup Script" to see how each one can be changed). Arguments
See also: Region Dataset, Sequence Partition, compare region datasets binding sequence occurrencesThis analysis can be applied to: Region Dataset
Description
This analysis is somewhat similar to the Count Motif Occurrences analysis, except that instead of just reporting the number of sites found for each motif (based on region type), the counts are further subdivided based on the sequence property of the motif site, which means that for each motif the analysis reports the number of sites found for each unique binding sequence. For example, if a motif with consensus "CAsGTG" occurs a total of 7 times, the analysis could report that it occurs 4 times with the specific binding sequence "CACGTG" and 3 times with the binding sequence "CAGGTG". For each combination of motif and specific binding sequence, the analysis reports how many occurrences there are in total of that binding combination, the number of sequences that contains this combination and also a match score for this combination. The match score is a relative score between 0 and 100 that reflects how well the specific binding sequence matches the motif. The best matching binding sequence (the one which gives the highest score according to the binding matrix) is given a score of 100 and the worst possible match is given a score of 0. Arguments
See also: count motif occurrences compare clusters to collectionNo documentation currently available.compare collectionsThis analysis can be applied to: Collection
Description
This analysis compares two collection objects (of the same type) to see if they have any entries in common. The analysis reports the number of entries that are present in both collections, in one of the collections but not the other and also the number of entries that not present in either of the two collections (but are present in a "total" collection). The analysis also calculates p-values which reflect the probability that the two collections should have at least the observed number of entries in common (or at most this number of entries in common) assuming the entries for the two collections had been randomly sampled from a larger collection (called "total"). Arguments
compare motif occurrencesThis analysis can be applied to: Region Dataset
Description
This analysis will count the number of times each type of motif occurs in one set of sequences (target set) and compare this to the number of times the motifs occur in a second set (control set). Statistical tests (either binomial test of hypergeometric test) will assess whether some motifs occur significantly more often in one of these sets compared to the other. The output contains counts of how many times each motif type occurs in the target set and control set respectively and also p-values for the target and control sets. These p-values reflect the probability of encountering the observed number of hits (or higher) given an expected number of hits based on each motif's frequency in the opposite set. E.g. If a specific motif occurs N times in the target set and M times in the control set, the reported "target p-value" will be the p-value of observing N or more motif hits in a dataset of the same size as the target set based on an expected motif frequency given by M divided by the size of the control dataset (or more accurately the maximum number of times a motif of that size could occur within such a dataset). Motifs that are significantly overrepresented in the target set are marked in red colors in the output, whereas those that are significantly overrepresented in the control set are marked in green. Motifs that are occur in both sets but are not significantly overrepresented in either set are marked with yellow. Arguments
See also: count motif occurrences, compare region occurrences compare motif track to numeric trackThis analysis can be applied to: Region Dataset and Numeric Dataset
Description
This analysis will compare a motif track against a numeric track and examine the numeric values found within each motif site. For each type of motif, the location of all binding sites (TFBS) for this motif are found. Next, different statistics are calculated based on the values that the chosen numeric track has within these TFBSs, including the smallest (minimum) value in the track within all TFBSs for each motif, the largest (maximum) value, the sum of all values within positions covered by TFBSs and the average value of the numeric track within the TFBSs (found by taking the sum and dividing by the total number of positions within the TFBSs). In addition, the analysis will also count the number of TFBSs for each motif where the average value of the numeric track within the TFBS (found by summing up the values within the TFBS and dividing by the length of the TFBS) is greater than (or equal to) some selected threshold. Arguments
compare region datasetsThis analysis can be applied to: Region Dataset
Description
This analysis compares two region datasets and calculates several ("nucleotide level") statistics based on their overlap, namely sensitivity (SN), specificity (SP), positive predictive value (PPV), negative predictive value (NPV), performance coefficient (PC), average site performance (ASP), F-measure (F), accuracy (Acc) and Matthews correlation coefficient (CC). See the "benchmark" analysis for a detailed description of these statistics. The formulas for all these statistics are based on four parameters that count the number of true positive nucleotides (TP), false positives (FP), true negatives (TN) and false negatives (FN) respectively. A true positive is a nucleotide position that is inside a region in both of the two Region Datasets. A false positive is a nucleotide position that is within a region in the first dataset but not in the second dataset. A true negative is a nucleotide that is outside regions in both datasets. A false negative is a nucleotide that is outside a region in the first dataset but inside a region in the second dataset. The analysis will also show a pie chart illustrating how much overlap there is between regions in the two datasets (fraction of nucleotides within regions in both sets), as well as the fraction of positions within regions that are unique to either the first or the second dataset and finally the fraction of nucleotides that are outside regions in both datasets ("background"). Arguments
See also: benchmark compare region occurrencesThis analysis can be applied to: Region Dataset
Description
This analysis will count the number of times each type of region occurs in one set of sequences (target set) and compare this to the number of times the regions occur in a second set (control set). A hypergeometric test will assess whether some regions occur significantly more often in one of these sets compared to the other. The output contains counts of how many times each region type occurs in the target set and control set respectively and also p-values for the target and control sets. Regions that are significantly overrepresented in the target set are marked in red colors in the output, whereas those that are significantly overrepresented in the control set are marked in green. Regions that are occur in both sets but are not significantly overrepresented in either set are marked with yellow. Arguments
See also: count region occurrences, compare motif occurrences count module occurrencesThis analysis can be applied to: Region Dataset
Description
This analysis counts the number of times each module occurs in a given module track (i.e. the number of sites for each module), and reports the total count for each module and also the number of sequences that contain each module. Arguments
See also: count motif occurrences, count region occurrences count motif occurrencesThis analysis can be applied to: Region Dataset
Description
This analysis counts the number of times each motif occurs in a given motif track (i.e. the number of binding sites for each motif), and reports the total count for each motif and also the number of sequences that contain each motif. If a Motif Numeric Map containing expected frequencies for each motif is specified (number of motif sites expected per position in the sequence), a p-value representing the probability of encountering at least as many motif instances as observed in the sequences will be reported and the statistical significance of motif overrepresentation will be assessed by a binomial test. Arguments
See also: count module occurrences, count region occurrences, compare motif occurrences, Motif Numeric Map count region occurrencesThis analysis can be applied to: Region Dataset
Description
This analysis counts the number of times each region type occurs in a given region track and reports the total count for each region type and also the number of sequences that contain each region type. For example, for a track containing repeat regions, the analysis will first determine which types of repeat regions are present in the sequences (e.g. different types of "Alu" repeats, SINEs, LINEs and simple repeats etc.) and then count the number of times each such repeat type occurs. Arguments
See also: count motif occurrences, count module occurrences evaluate priorThis analysis can be applied to: Region Dataset
Description
One of the key functionalities of MotifLab (and its predecessor PriorsEditor) is the ability to create numeric tracks that can be used as positional priors to guide motif discovery programs by assigning higher scores to positions that are considered more likely to harbour transcription factor binding sites. Such priors tracks can be created manually step-by-step by using different operations to combine information from multiple feature tracks or they can be generated automatically with PriorsGenerators that have been trained to discover the relationship between binding site occurrences and other genomic features. In either case, it will be useful to evaluate the potential of positional priors tracks generated in a certain way by comparing such a priors track against a region track containing known binding regions, to see if the track generated with this particular approach indeed has higher values inside these regions compared to outside. The "evaluate prior" analysis will do just this. The analysis has two different modes of operation depending on whether or not the optional "Priors track" parameter has been specified. If no "Priors track" has been selected, the analysis will be run in "general mode". However, if a "Priors track" has been selected, this particular track will be analyzed in more detail in "specific mode". General mode In general mode, all available numeric tracks and region tracks will be compared to the given target track and evaluated. For each track, a ROC-curve will be generated reflecting its potential for discriminating positions within regions from background positions based on the track's score at each position. Also, the area under the curve (AUC) will be calculated for the ROC-curve. The ROC-curve for a track is generated in the following way: First, all the positions within the track are sorted in ascending order according to the value at each position. Then, starting at (0,0) in the graph and going through the sorted positions one by one, the ROC-curve moves one step up if the next sorted position is within a region in the target track and one step to the right if the next sorted position is outside of any regions. After all positions have been covered, the ROC-curve should end up at coordinate (1,1). (Note that the ROC-graph has been normalized so that the x-axis represents the fractional number of positions that lie outside of regions and the y-axis represents the fractional number of positions that lie within regions). Hence, if a certain priors track tends to have higher values within regions of the target track compared to outside, the graph will tend to move more upwards at the beginning and then to the right at the end, resulting in a larger area under the curve. One the other hand, if a track tends to have higher values outside of the target regions, the graph will move to the right at the beginning and then more upwards towards the end, resulting in a smaller area under the curve. Higher AUC values thus means that the priors track tends to have higher values inside of target regions. If all positions inside of regions have higher prior values than the background (so a clear separation between regions and background can be made based on the priors values), the ROC-curve will move from (0,0) to (0,1) and then to (1,1) which gives a perfect AUC-score of 1.0. If a priors track tends to give equally high values to positions inside and outside of regions (so the positions inside and outside are about uniformally distributed when sorted by numeric value), the ROC-curve will tend to move in a straight diagonal line from (0,0) to (1,1) resulting in a AUC-score of 0.5. In this case, the numeric priors track shows no ability to discriminate between regions and background. ROC-curves for region tracks are calculated in a similar fashion by treating positions within regions as having a numerical value of 1.0 and positions outside regions as having the value 0.0. Note that even though ROC-curves and AUC-scores are calculated for all available numeric and region tracks, only the tracks that are currently visible in the GUI will be included in the graph whenever the analysis is displayed in a dialog or output using the "output" operation. Hence, if you only wish to include a few selected tracks in the graph, you can hide the tracks you don't want to include. Also, the color used for the ROC-curve of each track will be the same as the currently selected display color for that track. For analyses performed outside the GUI (running in CLI-mode from a protocol script), display setting statements can be used to hide tracks and set the colors for each track. Specific mode If a specific numeric track has been selected for the "Priors track" parameter, the analysis will be performed in "specific mode" which gives a more in-depth and detailed analysis of the potential of using the selected track as positional priors. First, the ROC-curve and area under the curve (AUC) is calculated for the priors track the same way as if the analysis had been performed in "general mode". Second, a "precision-recall" graph is calculated that shows the maximum "precision" (positive predictive value) that can be achieved for different recall (sensitivity) levels. The analysis will also produce additional graphs showing how the scores for several different nucleotide-level performance statistics will vary depending on a chosen cutoff threshold for the selected priors track. For a given threshold level, all positions where the value of the priors track is higher than or equal to this threshold (or strictly higher depending on the 'threshold' parameter) are considered "positive" positions and all positions with values below the threshold are considered as "negative". Positive positions that are within target regions are further classified as "true positives" (TP) and those outside are classified as "false positives" (FP). Conversely, negative positions inside target regions are classified as "false negatives" (FN) and those outside as "true negatives" (TN). These four parameters (TP/FP/FN/TN) serve as basis for calculating several nucleotide-level statistics that are described in detail in the manual entry for the benchmark analysis. For each nucleotide-level statistic, such as e.g. sensitivity, the threshold will be varied from the lowest numeric value in the priors track to the highest value (in increments of 1/100 of the range) and the graph will show the performance that can be achieved according to that statistic for each threshold level. For example, when evaluating "Conservation" as a priors track for predicting TFBS, the sensitivity value (y-axis) at threshold=0.65 (x-axis) reflects the fraction of TFBS positions that are correctly predicted if we assume that all positions that have a Conservation value of 0.65 or higher reside within TFBSs. The analysis will also determine two "optimal thresholds". The first is for the threshold value which gives the best trade-off between sensitivity and specificity (which is to say the threshold which results in the highest arithmetic mean of the sensitivity and specificity scores), and the second optimal threshold is the one which results in the highest possible score for the accuracy statistic. Arguments
See also: benchmark, numeric dataset distribution GC-contentThis analysis can be applied to: DNA Sequence Dataset
Description
This analysis calculates the GC-content (percentage) in a given DNA track for every sequence and possibly also additional statistics for a group or groups of sequences (such as the minimum, maximum, average and median GC-content for the sequences in the group). Arguments
See also: Sequence Collection, Sequence Partition motif collection statisticsThis analysis can be applied to: Motif Collection
Description
Calculates statistics related to motif size, IC-content and GC-content for the motifs in a given collection. The analysis reports the minimum, maximum, average, standard deviation, median and 1st and 3rd quartiles for these three motif properties and also shows histograms of their distributions. Arguments
motif position distributionThis analysis can be applied to: Region Dataset
Description
This analysis will analyze the positional distribution of each motif in a motif track. It can be used to assess whether motifs of certain types are uniformly distributed within sequences or if they tend to be located in the same location relative to a selected alignment anchor position across sequences (for example if some motifs tend to occur at the same distance relative to the transcription start site in several different sequences). To perform the analysis, the sequences are first aligned according to the selected anchor. Next, for each motif type the binding sites (TFBS) for this motif are located and a distribution is calculated based on the distance between the center of each TFBS and the alignment anchor. Different statistics can be calculated based on this distribution, but so far the only statistics reported are the standard deviation and kurtosis. In addition to these two statistics, graphical histograms can be created which shows the distribution of the binding sites for each motif type. Arguments
motif regressionThis analysis can be applied to: Region Dataset
Description
Arguments
See also: single motif regression motif similarityThis analysis can be applied to: Motif
Description
This analysis will compare a single selected motif against a collection of motifs using all motif similarity metrics that are known to MotifLab (which currently include "Average Log-Likelihood Ratio","Chi-squared","Kullback-Leibler Divergence","Pearson's Correlation","Pearson's Correlation (weighted)" and "Sum of Squared Distances"). The analysis will report the raw score values for these metrics. Arguments
numeric dataset distributionThis analysis can be applied to: Numeric Dataset
Description
This analysis will calculate distribution statistics for a Numeric Dataset; namely the number of bases in the track, the minimum and maximum values, the average and standard deviation and 1st, 2nd (median) and 3rd quartiles. If an optional Region Dataset is specified, the analysis will calculate separate statistics based on values inside regions in this dataset versus values outside these regions. A histogram will also be created showing the distribution of the values. In this histogram, the 1st/2nd/3rd quartiles plus the min and max values will be shown in a box-and-whiskers plot, whereas the average value will be shown as a diamond with standard deviation illustrated by arrows. Arguments
See also: numeric map distribution numeric map correlationThis analysis can be applied to: Numeric Map
Description
This analysis compares two Numeric Maps to determine if the values for corresponding entries are correlated (i.e. if entries that have relatively high values in the first map also have relatively high values in the second map, etc). The analysis calculates and reports two correlation statistics, namely "Pearson's correlation" and "Spearman's (rank) correlation" Arguments
numeric map distributionThis analysis can be applied to: Numeric Map
Description
This analysis will calculate distribution statistics for the values in a Numeric Map; namely the number of entries, the minimum and maximum values, the average and standard deviation and 1st, 2nd (median) and 3rd quartiles. If an optional Partition is specified, the analysis will calculate separate distribution statistics for each cluster in the Partition. A histogram will also be created showing the distribution of the values. In this histogram, the 1st/2nd/3rd quartiles plus the min and max values will be shown in a box-and-whiskers plot, whereas the average value will be shown as a diamond with standard deviation illustrated by arrows. Arguments
See also: numeric dataset distribution region dataset coverageThis analysis can be applied to: Region Dataset
Description
This dataset looks at the coverage of regions in a Region Dataset and calculates the fraction of each sequence that is covered by regions (in terms of nucleotides). It can also calculate min/max/average/median coverage statistics for a single group of sequences (specified as a Sequence Collection) or several groups of sequences (specified as clusters in a Sequence Partition). Arguments
See also: GC-content single motif regressionNo documentation currently available.ToolsMotifLab's graphical interface includes several tools that can be used to explore, analyse or manipulate data in an interactive manner. All tools can be found under the "Tools" menu in the main menu bar and some also have buttons in the tool bar.Actions performed with these interactive tools can not be recorded in protocols and can therefore not be repeated automatically (although some tools, like Crop/Extend Sequence, have analogous operations). Mouse toolsSelection tool
The Selection Tool can be used to select subsegments ("selection windows") of
your sequences and limit the application of some operations to positions
within these windows or regions overlapping the windows. To define a selection
window, point the mouse at either the start or end of the window within the
sequence, press the mouse button and drag the mouse to the other end of the
window. Selection windows will be shown as transparent yellow
overlays on the sequences. You can define several selection windows by holding
down the ALT-key while dragging the mouse (overlapping selection windows will
be merged). You can also subtract from the current selections by holding down
the SHIFT-key. If you click anywhere within a sequence without holding down
either ALT or SHIFT, the current selection windows will be discarded.
If you point the mouse at a sequence and press the A-key, the whole
sequence will be selected. If you press the I-key, the selection windows in
that sequence will be inverted. If you hold down the ALT-key while pressing
either A or I, this functionality will be applied to all sequences.
|
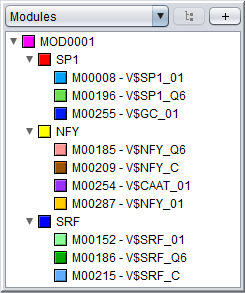
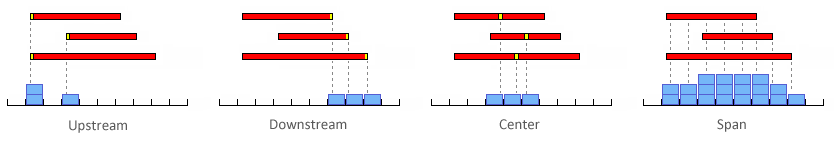
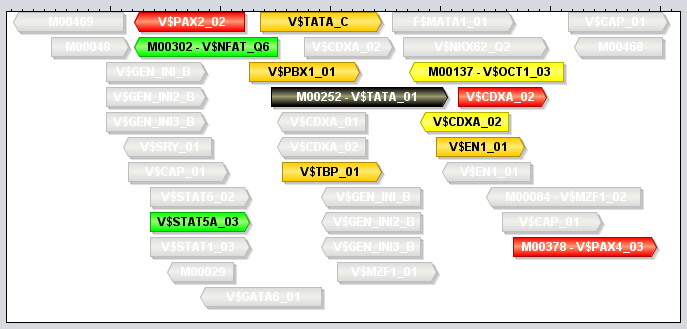
| Sort property | Effect |
|---|---|
| Sequence name | Sequences are sorted in natural order according to their names |
| Sequence length | Sequences are sorted by their lengths |
| Location | Sequences are sorted first by chromosome and then by position within the chromosome |
| (Visible) Region count† | Sequences are sorted according to the number of regions within each sequence with respect to a selected Region Dataset |
| (Visible) Region coverage† | Sequences are sorted according to the number of bases covered by regions within each sequence with respect to a selected Region Dataset |
| (Visible) Region scores sum† | Sequences are sorted according to the sum of region scores over all regions within each sequence with respect to a selected Region Dataset |
| Numeric map | Sequences are sorted according to their values in a selected Sequence Numeric Map |
| Numeric track sum | Sequences are sorted according to the sum of values within each sequence with respect to a selected Numeric Dataset |
| GC-content | Sequences are sorted according to their GC-content with respect to a selected DNA Sequence Dataset |
| Mark | This will place all marked sequences before unmarked ones when sorting in descending order |
The sorting algorithm is stable, so if you first sort by a secondary property (e.g. sequence name) and then by a primary property (e.g. Numeric Map value) the sequences that have the same primary property value (map value) will be sorted internally by the secondary property (name).
Group by Sequence Partition
This option will group sequences together into clusters according to a selected Sequence Partition. The sequences are first sorted by the name of their cluster and within each cluster the sequences are sorted by the chosen sort property.
Note that sorting options are also available from the context menu when right-clicking on a dataset in the Features Panel, and it is possible to sort sequences within a protocol script using the "sort(mode)=asc|desc" display setting command.
Crop Sequences
The Crop Sequences tool can be used to make sequences shorter by removing a number of bases from one or both ends of a sequence.All existing Feature Datasets will also be updated to conform to the new length.
Cropping can be performed in two different ways:
Removing a specific number of bases
This mode allows you to specify the exact number of bases to remove from the start and the end of the sequences respectively.
Note that which side of the sequence is considered the start or end depends on the strand orientation of each sequence
but also the selected orientation mode setting. With the "relative orientation" setting, the start of the sequence is the upstream end when oriented according
to the origin strand of the sequence. With the "direct orientation" setting, the start will always be the side with the smallest genomic coordinate and the end
the side with the largest genomic coordinate.
It is possible to crop a different number of bases from each individual sequence by specifying the number for each sequence
in a Sequence Numeric Map and then selecting this map as the argument in the dialog
rather than entering a constant number.
Provided with a region track, the sequences can be cropped so that the new start of the sequence corresponds with the start of the first region in the selected track and the new end of the sequence corresponds with the end of the last region. In other words, each sequence will be cropped so that it covers all regions present in the track but without additional flanking positions outside. Sequences that contain no regions at all will be left untouched rather than cropping them to 0 bp.
Cropping sequences can also be performed with the operation: crop_sequences.
Extend Sequences
The Extend Sequences tool can be used to make sequences longer by adding new base positions to one or both sides of a sequence.The tool takes two numeric arguments specifying the number of bases to add to the start of each sequence and the end of each sequence respectively. Note that which side of the sequence is considered the start or end depends on the strand orientation of each sequence but also the selected orientation mode setting. With the "relative orientation" setting, the start of the sequence is the upstream end when oriented according to the origin strand of the sequence. With the "direct orientation" setting, the start will always be the side with the smallest genomic coordinate and the end the side with the largest genomic coordinate.
It is possible to add a different number of bases to each individual sequence by specifying the number for each sequence in a Sequence Numeric Map and then selecting this map as the argument in the dialog rather than entering a constant number.
MotifLab is not able to extend existing Feature Datasets associated with the sequences (since it does not necessarily know the values for these datasets outside the current range and by design refuses to fill in with blanks). Because of this, sequences can only be extended as long as no Feature Datasets are present.
Extending sequences can also be performed with the operation: extend_sequences.
Other Tools
Update Motif Properties
Documentation in preparationConfiguring MotifLab
General options
The general configuration options for MotifLab can be edited in the Options dialog, which can be accessed under "Options..." in the "Configure" menu in the main menu bar. The dialog organizes the options under different tabs.General
Concurrent Computational Threads
The number of concurrent computational threads can be increased to allow MotifLab to take advantage of parallel processing on computers that have multiple cores (which most computers have these days). Note that this functionality is not fully utilized in the current version of MotifLab (but is used by e.g. the SimpleScanner motif scanning program).Maximum Concurrent Downloads
This setting specifies how many concurrent download request Motiflab can make to the same server. If this value is set to 1, MotifLab will always wait for any requested file to be completely downloaded before making a new request to the same server. If a value X higher than 1 is specified, MotifLab will have a pool of X connections open simultaneously. Each connection can make a new request to the server as soon as the previous file requested on that connection has been completely downloaded. Allowing more concurrent downloads will normally result in faster download times, but it will also put more strain on the servers (which could potentially result in users being banned from connecting to particular servers).Network Timeout
The network timeout setting specifies the amount of time (in milliseconds) that a server contacted by MotifLab has to respond before a "network timeout" error will be reported.Maximum Sequence Length
MotifLab has primarily been designed to perform operations and analyses on multiple, short sequence segments rather than very long (e.g. genome-wide) sequences. The Maximum Sequence Length setting can be used to safeguard against accidentally specifying overly long sequences in the Sequence Dialog (which could for instance happen if a user types a digit or two too many for the end coordinate of a sequence compared to the start coordinate), as this could result in the system being bogged down while attempting to download an excessive amount of data for this sequence region.TSS at position
In the bioinformatics community it is common to refer to the first base in a gene sequence (the TSS) as position "1" and the second base as "2" (and so on), while the first position upstream of the gene is referred to as "-1". A number line with gene-relative coordinates thus goes directly from the negative numbers (for positions upstream of the gene) to the positive numbers (for positions inside the gene), hence skipping the zero-position. The TSS at position setting can be used to specify whether this particular convention of going directly from -1 to +1 should be followed (by selecting "TSS at position +1") or whether the zero-position should be included (by selecting "TSS a position +0") as with a regular number line.Note that this setting is used by the Sequence Visualizer (and its ruler and tooltips) but is not necessarily respected by other parts of the system, such as Data Formats that output Region Datasets using TSS-relative coordinates. However, these formats usually have their own parameters that can be used to specify if the +0 position should be skipped.
Autocorrect Sequence Names
This option was introduced in MotifLab version 2.0. MotifLab requires the names of sequences to only consist of letters, numbers and underscores. However, some sequence identifiers, for instance in yeast, can contain other characters as well (hyphens in particular). If the "Autocorrect sequence names" option is selected, MotifLab will automatically convert illegal sequence names to legal sequences names (usually by replacing illegal characters with underscores) whenever data is read from files.Ask Before Discarding Data
If this option is selected, MotifLab will display a popup dialog whenever one of the "Clear Data" functions is selected from the "Data" menu. The dialog will ask the user to confirm that they really would like to delete the data objects and allow them the chance to change their mind. Also, if a user closes a protocol in the protocol editor or closes an output panel containing contents that has not been saved, MotifLab will ask the user if they would like to save the document before closing it. Note that data that is deleted with the "delete" operation or by selecting a data object and pressing the DELETE key will not be affected by this setting.Save Session On Exit
This option was introduced in MotifLab version 2.0 and can be set to either "Always", "Ask" or "Never". When set to "Always", MotifLab will always save the current session (to an internal file) when the program is exited and restore this session automatically the next time MotifLab is started. If set to "Ask", MotifLab will display a popup dialog when the program is about to exit which allows the user to choose whether or not to save the current session (and restore it next time). Setting this option to "Never" will disable the auto-save/restore functionality.Visualization
The "Visualization" tab contains options for configuring how sequences and feature data tracks are displayed in the main visualization panel.Sequence Window Size
This option will set the width of the sequence windows (displayed tracks), and this can be useful to adjust if you have a computer screen which is either smaller or larger than assumed by the default setup.Sequence Label Size
This option will set the width of the sequence labels displayed in front of the sequence windows (data tracks). This width can either be set to a fixed size (in pixels) or to a size which is determined by the system based on the size of the label for the sequence with the longest name. The latter option is enabled by checking the scale to fit box (recommended). If a fixed label size (not "scaled to fit") is used and the length of a sequence label is larger than the specified size, the label will be drawn on top of the sequence window (thus obscuring the data tracks displayed underneath).Antialias text and Antialias motif logos
These two options can be used to turn on or off anti-aliasing on motif logos and text displayed in the sequence window (such as sequence labels, numbers and labels in the ruler and coordinates in the info-panels). Enabling anti-aliasing will allow fonts to be rendered more smoothly than if antialiasing is turned off, which makes the graphics more aesthetically appealing but also easier to read (especially for small font sizes). However, on older computer systems, anti-aliasing would have a performance penalty, which is why these two options were included to turn it off.Background color
This setting can be used to change the color of the background in the visualization window. The button named "Color" will be displayed in the currently chosen background color, and clicking this button will display a pop-up dialog which allows the user to select a different background color. Clicking the "Reset" button will revert the background color to the default setting (which will be a gray color).Cache
The "Cache" options tab allows users to turn on or off caching functionality and also clear all the contents of the caches.Obtaining feature data from external servers could potentially take a long time, which is why MotifLab has options to locally cache data that has been downloaded. Whenever a user requests to obtain data for a certain feature, MotifLab will first check if all or part of the requested data is already available from the local cache, and it will only make connections to external servers in order to obtain data that can not be found in the cache. Also, when users rely on different types of gene identifiers to define which sequence regions to work on, MotifLab will have to contact an external service (usually BioMart) to resolve these gene IDs and determine the genomic coordinates of these sequences. The mapping between gene identifiers and gene locations can also be cached so that this information is readily available for sequences that have been analyzed before. When caching is turned off, no new information will be stored in the cache and MotifLab will not make use of any data that might be present in the cache from before. Turning off caching will not destroy any data presently in the cache, however, so reenabling caching will give access to all data that was previously cached.
Protocol Editor
The "Protocol Editor" options panel can be used to set the colors used for coloring keywords in protocols. The panel contains several colored buttons, including e.g. "Operations", "Data objects", "Data Formats" and "Numbers", that refer to different classes of keywords. The color of each button reflects the color currently used for keywords of that class. To change the color for a particular class, simply press the corresponding button and select a new color from a pop-up dialog.HTML
The "HTML" panel contains a few options relating to output files in HTML format produced by MotifLab's output operation. HTML documents rely on Cascading Style Sheets (CSS) to define a style for the document (affecting e.g. fonts and colors) and also use JavaScript to enable certain interactive functionality, such as sorting tables by clicking on a column header. The HTML options specify how the required style information and JavaScript code should be made available to the HTML documents.| New File | For each HTML document in MotifLab that is saved to file, new separate file(s) will be created to hold the style information (or JavaScript code) and these files will be referenced by name from within the HTML-file. A new CSS-style (or JavaScript) file is saved to the same directory as the HTML-file and the name of the file will be the same as the HTML-file except that the suffix is changed to ".css" (or ".js") rather than ".html". Using a new style- or javascript-file for each HTML document allows different documents in the same directory to have different styles and functionalities. |
| Shared File | When this option is selected, the style sheet or JavaScript code is output to a file with a fixed name ("motiflab_style.css" and "motiflab_script.js"), and all HTML documents that are created will contain references to these files (which are assumed to reside in the same directory). When a new HTML-file is saved to a directory which does not contain these files, they will be created. However, subsequent HTML documents saved in the same directory will simply rely on the same files. This means that you can easily change the style or JavaScript functionality for all the HTML-files residing in the same directory simply by editing or replacing the "motiflab_style.css" and "motiflab_script.js" files. |
| Embed | For each HTML document created, the code for the CSS style or JavaScript required will be included in the HTML document itself. This means that the HTML documents will be self-contained since they do not rely on other external files (at least if both CSS and JavaScript is embedded). |
| Link | When this option is selected, HTML documents will link to stylesheets and JavaScript files residing on the MotifLab web server. This means that no new stylesheet or JavaScript files are created locally when HTML documents are saved to file, but access to the MotifLab web server would be required in order to display these HTML files properly in a web browser (they can be displayed with default style and no JavaScript functionality, however). Note that linking does not work for CSS style sheets prior to version 2.0 of MotifLab due to a bug. |
| None | This setting specifies that no CSS style information (or JavaScript) should be associated with the HTML document. This would mean that a default style should be used or that functionality requiring JavaScript should be disabled. |
The Stylesheet option is used to select which CSS stylesheet to use for HTML documents. Users can choose between a few predefined styles installed with MotifLab or select a homemade CSS-file. To use a predefined style, the name of the style should be typed in brackets in the stylesheet box (e.g. "[green]"). As of MotifLab v2.0, only two predefined styles are available: default and green.
Configuring external programs
XML configuration files for external programs
In order to use external programs within MotifLab, their interfaces must be explained to MotifLab through special configuration files written in an XML-format which is explained below. If you want to check out more examples you can have a look at the configuration files for various supported programs available from the external programs page.Example:
The box below shows the XML-code required to configure a simple program called "randomfilter.exe" which takes three arguments: the name of a GFF-formatted input-file, a number between 0 and 1.0, and the name of an output-file. The program would read the GFF-file line by line and with a given probability write the line to the new output-file. The command to execute this program from a CLI-shell would be
<?xml version="1.0" encoding="UTF-8"?> <program name="RandomFilter" class="Filter"> <service type="local" location="C:\bioinformatics\randomfilter.exe" /> <parameter type="regular" name="Region Track" class="RegionDataset" required="yes"> <dataformat name="GFF" /> <argument type="valued option" switch="-i"/> </parameter> <parameter type="regular" name="Probability" class="Double" required="yes"> <min>0</min> <max>1.0</max> <default>0.5</default> <argument type="valued option" switch="-p"/> </parameter> <parameter type="result" name="Result" class="RegionDataset" required="yes"> <dataformat name="GFF" /> <argument type="valued option" switch="-o"/> </parameter> </program> |
After the compulsory XML-header in the first line follows a <program> element which contains the actual description of the program and its interface.
The <program> element has two arguments: a name and a class. The name argument is just a name selected to refer to the program.
The class argument tells MotifLab what kind of program this is. Five special classes are recognized which can also have specified requirements on the configration file. These are "MotifDiscovery", "MotifScanning", "ModuleDiscovery", "ModuleScanning" and "EnsemblePrediction". Programs from these five classes are executed with corresponding operations in MotifLab; e.g."MotifScanning" programs are executed with the "motifScanning" operation, etc. For programs that do not fall within the special classes, the class argument is merely descriptive and can be set to any value. For example, since the "randomfilter" program above is used to filter data, it is given the arbitrary value "Filter" for the class argument. Programs that are not one of the special classes can be run with the "execute" operation.
A third and optional argument to <program> is cygwin which can take on the values "yes" or "no" (default is "no"). This argument can be used to signal that the program is originally a UNIX/LINUX program and needs Cygwin to be installed in order to run under WINDOWS operating systems. If cygwin is set to "yes" some filepaths might be converted to UNIX-style as necessary.
The <program> element further contains other elements that describe various properties of the program, including information about where the program is located, how to execute it and descriptions of the input and output parameters of the program.
Program properties
The <program> element can contain an optional <properties> element which describes various properties of the program, including names of the authors, a short description of the program itself, contact information, websites and citations. These properties are displayed in the HELP-page for the program (which is shown for instance when the user double-clicks on a program in the External Programs Dialog), and they are mostly useful if one wants to share an XML-configuration file with other users that are not familiar with the program. The <properties> element can also contain a <license> element with a license agreement that the user must accept in order to use the program and a <register> element containing a web address where the user can be directed in order to register their use of the program. HTML-code can be used in the text of these elements as long as the angle brackets used around HTML-elements are escaped (for example, to use italics, "<i>" must be escaped as "<i>").
<properties>
<author>Timothy L. Bailey and Charles Elkan</author>
<citation>
Timothy L. Bailey and Charles Elkan (1994)
"Fitting a mixture model by expectation maximization to discover motifs in biopolymers",
<i>Proc 2nd Int Conf on Intelligent Systems for Molecular Biology</i>,
(28-36), AAAI Press, 1994
</citation>
<contact>donotreply@somewhere.org</contact>
<homepage>http://meme.sdsc.edu</homepage>
<description>
MEME searches for novel motifs in DNA (and protein) sequences
using an expectation maximization strategy
</description>
<register>http://www.server.org/software/register.cgi</register>
<license>
In order to use this program you must agree not to use it for commercial purposes
</license>
</properties>
|
Service type and location
The <service> element describes the program's location and how it should be accessed. The current version of MotifLab only supports use of programs that are installed locally on the user's computer (type="local"), but future versions might also support the use of web services. (The special setting type="bundled" is used for programs that come shipped with the installation of Motiflab). If the location of the executable program is known, it can provided as an argument to the <service> element, as seen in the example for the "RandomFilter" program on top of this page. If the location of the program is not stated in the XML-file, the user must specify the location when the XML-file is installed in MotifLab. If a precompiled executable of the program can be obtained from an external source such as a web server, the location of this source can be provided inside the <service> element using optional <source> elements. The version and os arguments just provide a description for the program source, but the url argument must point to a single file that can be downloaded and "installed" locally by MotifLab. The downloaded file must be executable and usable "as is" since MotifLab is not capable of performing any special installation steps that the program might require. The only processing MotifLab can do is to unzip a program contained within a ZIP file. In this case the argument compression="ZIP" must be set (as shown for the second source below) and the location of the executable file within the ZIP archive must be specified with the targetInZIP argument.Version 2.0 of MotifLab introduced the require element which can be used to inform the user that this program or configuration file has certain system requirements, for instance that a certain version of JAVA must be installed or that this particular configuration file is only meant to be used with version X of the program in question. These requirements will be shown to the user when the program is configured in MotifLab. A special requirement is "MotifLab version X" which says that this configuration file will only work with a certain version of Motiflab (or more recent versions) since the configuration relies on functionality that is not present in earlier versions. If such a requirement is specified, a user will not be able to configure the program unless the required MotifLab version is used.
<service type="local">
<source version="3.1" os="Windows"
url="http://homes.esat.kuleuven.be/~thijs/download/windows/MotifScanner.exe" />
<source version="3.1" os="Windows (mirror)"
url="http://tare.medisin.ntnu.no/priorseditor/tools/windows/MotifScanner.zip"
compression="ZIP" targetInZIP="bin/MotifScanner.exe" />
<source version="3.2" os="Linux"
url="http://homes.esat.kuleuven.be/~thijs/download/linux_3.2/MotifScanner" />
<source version="3.2" os="Linux x86-64"
url="http://homes.esat.kuleuven.be/~thijs/download/linux_x86-64/MotifScanner" />
<source version="3.2" os="Mac OS X"
url="http://homes.esat.kuleuven.be/~thijs/download/macosx_ppc/MotifScanner" />
<require>MotifLab version 2.0</require>
<require>Java version 1.7</require>
</service>
|
Describing the program's interface
The description of the program's command-line interface mostly consists of a list of <parameter> elements, each describing an input or output parameter of the program.
<parameter type="regular" name="Positional priors" class="NumericDataset"
required="no" hidden="no">
<description>
A positional priors track (Note: sum of priors for all positions must not exceed 1.0!)
</description>
<argument type="valued option" switch="-psp"/>
<dataformat name="PSP">
<setting name="Orientation" class="String">Direct</setting>
<setting name="Motif width" class="Integer">8</setting>
</dataformat>
</parameter>
|
Each parameter has a type argument which can be either "source", "result" or "regular". Source parameters refer to existing data objects that are passed on to the external program for processing and result parameters refer to results output by the external program that are read back and converted into new data objects by MotifLab. The five special classes of external programs (motif/module discovery/scanning and ensemble programs) have specific requirements on the number and roles of source and result parameters. For example, motif scanning programs must have exactly one source parameter representing the DNA Sequence track and one result parameter (which must be called "Result") referring to the Region Dataset (motif track) returned my the motif scanning program. Motif discovery programs on the other hand must have two result parameters, which must be called "Result" and "Motifs" respectively, which refer to the motif track and motif collection objects returned by the motif discovery program. Programs can have additional parameters settings besides the input and output parameters which can be used to modify the behaviour of the program. These are then specified as "regular" parameters. Note that "source" parameters are only used by the five special program classes, and other classes should use "regular" parameters also when referring to any data passed on to the external program.
In addition to a type, a parameter must also have a name argument, which is used to refer to the parameter and is also the name displayed in GUI dialogs. Finally, a parameter must have a class argument which specifies the type of data the parameter holds. The class argument can refer to one of the four "basic types" String, Integer, Double and Boolean (for backwards compatibility Double can also be referred to as Float) or to one of MotifLab's own data types. Such data types must then be written without spaces and in camel case (where each "word" begins with a capital letter), such as for instance RegionDataset, MotifCollection and SequenceNumericMap. For Numeric Maps it is also possible to append a plus-sign to the class name. This then taken to mean that a Numeric Variable or literal numeric constant can be chosen by the user instead of a Map when selecting a value for the parameter (e.g. "MotifNumericMap+").
Parameters can have additional optional arguments such as: required, advanced, hidden and skipIfDefault which can be set to either "yes" or "no". Required parameters must be assigned values, and MotifLab will not allow a user to execute a program before he or she has chosen values for all required parameters (non-required parameters can be left blank and rely on defaulting values). Advanced parameters will not be shown in the GUI unless the user explicitly selects to display them by pressing a "+" button. If a program has many parameters, this option can be used to show only the most important parameters and hide the less frequently used parameters (which usually rely on defaulting values anyway) in order to make the visual presentation of the program's settings more tidy. Although not required, it is recommended that all advanced parameters be listed after the non-advanced parameters. Hidden parameters do not show up in GUI dialogs at all, and the user can not change the value of a hidden parameter directly. Hidden parameters can, however, be used to pass default settings to programs and they can also be indirectly updated in a preconfigured way in response to user selections. Arguments that have the skipIfDefault setting on will not be included on the command line if the parameter has the default value (which can be no value for non-required parameters). Unless these optional arguments are specified their default settings will be required=yes, hidden=no, advanced=no, skipIfDefault=yes.
<parameter> elements can contain other elements, for instance an optional <description> of the parameter which can be displayed to the user in a GUI dialog (HTML-code can be used if angle brackets are escaped as explained above).
The <argument> element inside the parameter is required and describes how the parameter is passed to the program. The argument can specify a switch which will preceed the parameter on the command line. Programs that rely on switches usually allow the parameters to be listed in any order on the command line since the switches can be used to identify the parameters. On the other hand, for programs that do not rely on switches, the parameters must be listed in a specific order to correctly interpret the command line. The argument element must specify a type which can be either "valued option", "flag", "explicit", "implicit", "STDOUT" or "STDIN". (The "explicit" type was introduced in MotifLab v2.0.)
Valued option parameters are those that pass some kind of value along to the program. Basic values, such as numbers, simple text strings or Booleans will be output directly on the command line. More complex data objects, on the other hand, will be written to temporary files (in specified file formats) and the name of the file will be referenced on the command line instead. The filename will normally just be some random (but unique) name chosen by the system. However, when it is necessary to use a particular filename, the argument type can be set to explicit rather than valued option and the filename can then be explicitly specified (similar to the last "implicit" parameter in the example below). Flag parameters are used for boolean settings. If the option related to a flag-parameter is selected, the parameter's switch will be output to the command line. If the option is not selected, the parameter will not show up on the command line at all. An implicit parameter will be tied to a specific value which is fixed and already known in advance. The value of this parameter will thus not depend on any current settings selected by the user. Implicit parameters can for example be used to refer to an output-file created by the external program when the name of that file is always the same and not chosen by the user. Some programs will read their input data from STDIN rather than a regular file and/or write output to STDOUT instead of a regular file. The special type values "STDIN" and "STDOUT" can be used to signal that a parameter relies on these standard streams rather than regular files. These types can thus be considered as special cases of implicit parameters. Note that a program can only refer to one STDIN and one STDOUT parameter per command element (explained below).
Example:
The following configuration file is for a program called "scan.exe" which requires a DNA file (in FASTA format) as its first input argument. It is also possible to specify two additional optional arguments, one which specifies a background model (preceeded by the "-b" switch) and one which tells the program to scan the reverse strand rather than the direct strand ("-r" switch). The program then outputs its results to a GFF-file called "output.gff" (this name is hardcoded in the program and is not possible to change).
The command to execute this program from a CLI-shell would then be
<program name="Scan" class="scanning">
<service type="local" location="C:\bioinformatics\scan.exe" />
<parameter type="regular" name="DNA" class="DNASequenceDataset" required="yes">
<dataformat name="FASTA" />
<argument type="valued option"/>
</parameter>
<parameter type="regular" name="Background" class="BackgroundModel" required="no">
<dataformat name="PriorityBackground" />
<argument type="valued option" switch="-b" switchseparator=" " />
</parameter>
<parameter type="regular" name="Scan reverse strand" class="Boolean" required="no">
<argument type="flag" switch="-r"/>
</parameter>
<parameter type="result" name="Result" class="RegionDataset">
<dataformat name="GFF"/>
<argument type="implicit" filename="output.gff"/>
</parameter>
</program>
|
Note that the configuration file specifies four parameters but the command line only has three parameters. This is because the last "result" parameter which captures the output from the program refers to a file which is implicit rather than being explicitly mentioned on the command line. When the command line to run this program is created, the parameters will be included in the order they are listed in the configuration. Because of this, the parameter referring to the FASTA file, which the program expects to be the first argument on the command line, must also be the first parameter in the configuration (a later section will describe a different way to construct the command line which foregoes this requirement). The first parameter (called "DNA") refers to a DNA Sequence Dataset object selected by the user. Since this parameter has the "valued option" argument-type, the selected data object will be output to a file (in the FASTA-format specified by the <dataformat> element) and the filename will be included on the command line. (If the class of the parameter had been either Integer, Double, Boolean, String or Numeric Variable its value would have been included directly on the command line).
The second parameter ("Background") is not required and will only be included on the command-line of the user has explicitly selected a Background Model for this parameter. In this case, the Background Model object will be written to a file in "PriorityBackground" format and the filename will be added to the command line after the parameter's specified switch, which in this case is "-b". The optional switchseparator specifies a string used to separate the switch from the parameter's value (in this case the name of the background file) on the command line. The switchseparator defaults to a single space, but is is also possible to specify other separators, for example a colon or an equals sign (in which case the parameter would appear on the command line as "-b:somefilename.bg" or "-b=somefilename.bg").
The third parameter ("Scan reverse strand") refers to a Boolean setting (these are usually displayed as checkboxes in the GUI). Since the argument-type in this case is set to "flag", the switch specified for this parameter ("-r") will only be added to the command line if the Boolean value is TRUE.
The fourth and final parameter ("Result") is a result-type parameter, which means that MotifLab expects to read some file that has been produced by the external program and use the information therein to create a new data object — which in this case should be a Region Dataset. As specified, the file should be in GFF-format. Also, since the argument-type of this parameter is set to be "implicit" the name of this output file is not referenced on the command line. Rather, the filename is specified directly.
Restricting values of simple parameters
Simple parameters such as Integers, Doubles, Strings and Booleans can be given default settings with a <default> element inside the parameter, as can be seen in the example on top of the page for the second parameter (Probability). For number parameters the allowed range can also be specified by providing <min> and <max> elements (although this is not checked in the current version of MotifLab). String parameters can normally take on any value, but they can also be restricted to a limited set of options:
<parameter class="String" name="Size" type="regular" >
<option>Small</option>
<option>Medium</option>
<option>Large</option>
</parameter>
|
<parameter class="String" name="Size" type="regular" >
<option value="S">Small</option>
<option value="M">Medium</option>
<option value="L">Large</option>
</parameter>
|
Specifying the data format for complex parameters
Complex parameters (not simple numbers, Strings and Booleans) are passed to external programs via temporary files. In order to output these parameters to files, the data format to use must be specified with a <dataformat> element inside the parameter. The name of the format must be given and the format might also require specification of additional format-specific <settings>. Each setting has a name and a class class (similar to the class of parameters as described above). Since the data format settings used by an external program is normally decided in advance and hence fixed, the values for the settings are usually constant values written between the <setting> and </setting> tags. However, it is also possible to dynamically set a value using a link to another previously defined parameter (of the same class). For example, the "PSP" data format below specifies values for four settings (if the PSP format had other settings these would take on default values). The first three settings have fixed values, whereas the last setting "Motif width", which is an integer number, takes its value from another parameter called "Motif Size" (which should be an integer-class parameter that has been defined earlier in the XML-file). Please consult the Data Formats section of the user manual for detailed descriptions of each particular data format and their settings.
<parameter type="regular" name="Positional priors" class="NumericDataset"
required="no" hidden="no">
<dataformat name="PSP">
<setting name="Orientation" class="String">Direct</setting>
<setting name="Normalize"class="String">Max 1</setting>
<setting name="Include width" class="Boolean">true</setting>
<setting name="Motif width" class="Integer" link="Motif Size" />
</dataformat>
</parameter>
|
Setting up the command line
The command line used to execute the external program can be defined in two different ways. One way is to explicitly specify the command-line, using the <command> element as described below. This method is the most powerful. However, programs that have very straightforward interfaces can do without the command-element.If no <command> element is specified, the command-line is build up by writing out the name of the executable program followed by all the parameters in the order that they appear in the XML-file. The values of "simple" parameter types, like numbers and strings are written directly to the command-line whereas complex types (such as large datasets) are written to temporary files and the filename is written to the command line. If a parameter has an associated switch then the switch is written out before the parameter itself. If the parameter is a boolean "flag", only the switch is output (or not, depending on the boolean value of the parameter). "Implicit" arguments are not written to the command line, however. Implicit arguments can be used when the value for a parameter is always the same, for instance if the external program always writes its output to a file named "output.txt" which is not referenced on the command line. Arguments that are implicit should specify the (already known) filename instead of a switch (unless they link to other parameters).
If the "RandomFilter" program described at the top of this page is executed, and the user has chosen a region dataset to use for the first parameter and a value of e.g. "0.45" to use for the second parameter, the resulting command-line that is executed will look like this:
C:\bioinformatics\randomfilter.exe -i <tempfile_1> -p 0.45 -o <tempfile_2> |
Before executing the command, however, the region dataset the user selected for the first regular parameter is output (in GFF-format) to a temporary file named tempfile_1. The third parameter also refers to a region dataset, but since this is a "result" parameter only the name of the file (randomly chosen for the occasion) is passed to the external program on the command-line. The external program is expected to write its output to this file (in GFF-format as specified in the XML-file) whose contents will later be read back by MotifLab after the program execution has finished.
The command element
If the program requires a more complex command-line than just the name of the program followed by the parameters in the order specified, the command-line can be specified explicitly with a <command> element. For instance, if the RandomFilter program above was not a standalone executable, but rather a perl script, we might have to specify the command-line like this.
<command>perl %PROGRAM {Region Track} {Probability} {Result}</command>
|
Here, %PROGRAM is a special string which refers to the program itself (this was implicit when we didn't use the command-element). Other special strings that can be used include %APPDIR which refers to the directory where the program resides, and %WORKDIR which is the "working directory" used when executing the command. Parameters are referred to on the command line by placing the name of the parameter in braces. The command-line will parsed and these braces will be replaced by the actual value of the parameter (or a filename for complex parameters) possibly preceeded by a switch if one is specified.
It is possible to specify multiple commands that should be executed in succession. This can be useful for instance if there is a need to perform any pre- or post-processing steps before or after running the program itself (for instance to convert output in a non-standard format produced by the program to GFF which can be read by MotifLab). There are two ways to specify multiple commands. The simplest way is to just include multiple commands in the same <command> element and separate those commands with a semicolon. Since some programs or operating systems might use semicolons for other purposes on the command line (for example to separate multiple paths in a JAVA classpath), it is possible to specify alternative characters (or even strings) to separate the commands via a separator argument to the command element. For example, the line <command separator="#"> uses the # sign to separate commands rather than the default semicolon.
The second way to specify multiple commands is to include a list of <command> elements. Note than in order to use this option, this list must be enclosed in an outer <commands> element to signal that the commands belong together.
<commands>
<command> first command... </command>
<command> second command... </command>
<command> third command... </command>
</commands>
|
An XML-configuration file should preferably be designed to be usable irrespective of which operating system the program will eventually run on. However, references to specific files within a command line might be tricky since different operating systems have different ways of representing file paths. Also, some operating systems might need to escape filenames containing spaces by enclosing them in quotes. MotifLab performs the necessary conversions automatically for temporary files and the %PROGRAM special string, but if you want to refer directly to other files within the command line, you might have to explicitly state that this part of the string refers to a file and should be processed accordingly. There are two ways to inform MotifLab that you want to refer to a file, and both work by enclosing the filename in "special quotes". The first uses "dollar-brace" style, like this: ${filepath}$, and the other uses "dollar-quote-brace" style, like so: $'{filepath}'$. (Note that the closing parenthesis is the reverse of the opening parenthesis). The difference between these are really only apparent for programs that run on WINDOWS using CYGWIN. With the first style, WINDOWS-paths are converted to CYGWIN Unix-style paths and enclosed in quotes if they contain spaces. The latter style does not convert the paths but will enclose them in quotes if they contain spaces. Use the latter style to refer to programs that should be executed and the first style for other file references. For an example of usage of the latter style you can have a look at the XML-configuration file for Weeder.
Sometimes, different operating systems can have a totally different command line syntax for the same program. To cope with such cases, you can specify a different command element for each operating system and use the os argument of the command to tell MotifLab which operating system the command pertains to, like so <command os="windows">. The "windows" string can be used to refer to all versions of windows, but for other operating systems the OS-string should match the (case-insensitive) String that will be returned by a call to the JAVA method System.getProperty("os.name"). The os argument also applies to the <commands> element used to group together multiple commands, so you can have different command groups for each OS. If no OS is specified for a command, it will apply to all operating systems and act as the default if no other more specific commands apply (e.g. if a configuration file contains two command elements, one with os="windows" and one with no OS argument, systems running windows will use the windows-specific command and all other systems will use the other command).
There may be cases when programs behave so differently on different operating systems that a simple rephrasing of the command line to execute the program is not sufficient to make the configuration compatible with multiple systems. It could be, for example, that a program has substantially altered functionality depending on the OS, uses different parameters or relies on other data formats for input and output. In such cases the system element can be used to group together elements that apply to different operating systems. A system element should be a direct child of the program element and can contain command, parameter, report and temporary elements that are specific to an operating system. Just like the command element, each system should also have an os argument which specifies which operating system it applies to (and a system element without such argument applies to all operating systems for which no other more specific system element is found).
Linking to other parameters
It is possible for a parameter to take on the same value as another parameter by "linking" to this other parameter. This is accomplished by specifying a link argument containing the name of the target parameter. Note that parameters can only link to other parameters that have already been defined earlier in the XML-file and they can only link to parameters of the same class. Parameters (except result parameters) that link to others should be "hidden", since their values should not be explicitly set by the user (only indirectly via the parameter being linked to). Settings for data formats can also link to other parameters (but not other settings) as explained above, and this is the only way a user can (indirectly) change values for data format settings (since information about data formats used for passing parameters is not usually revealed to the user).For example, motif discovery programs require two result-type parameters to be defined called "Results" and "Motifs" which will hold respectively the binding sites and motifs discovered by the the program. Each of these parameters is processed individually by MotifLab since the data produced for each parameter could potentially be output to different files by the program (the MotifSampler program for example outputs one GFF-file containing the prediced binding sites and one file containing the motif PWMs). However, many programs output all their results to a single file, and this will require both of these parameters to reference the same file (and usually this also means that a new program-specific parser has to be included in MotifLab). The code below shows these two result parameters defined for a hypothetical motif discovery program ("ProgramX") which allows the name of the single output file to be given on the command line using the switch "-o <outputfile>. The parameter defined first ("Result") references the file on the command line directly by using a "valued option" argument. The second parameter ("Motifs"), however, references the same file by linking to the first parameter (and declaring itself an "implicit" argument). It would be possible to use two different data formats for parsing the results file, one for each parameter. However, the solution below uses the same data format ("ProgramXFormat") for parsing both the binding sites and the motifs in the same file. Instead, the data format-specific setting "Parse" (which can here have the value "Sites" or "Motifs") is used to tell the ProgramXFormat which parts of the information in the file it should concentrate on and also what data it should return to MotifLab.
<parameter type="result" name="Result" class="RegionDataset">
<argument type="valued option" switch="-o" />
<dataformat name="ProgramXFormat">
<setting name="Parse" class="String">Sites</setting>
</dataformat>
</parameter>
<parameter type="result" name="Motifs" class="MotifCollection" link="Result">
<argument type="implicit" />
<dataformat name="ProgramXFormat">
<setting name="Parse" class="String">Motifs</setting>
</dataformat>
</parameter>
|
Parameters that link to other parameters will either reference the same atomic value as the target parameter (for the basic types Integer, Double, Boolean and String) or reference the same file as the target parameter (for all other complex data types). However, sometimes it could be necessary for a complex-type parameter to reference the same data object as another parameter but to have this object output to a different file in a different format. This can be accomplished by declaring the parameter to be a softlink rather than a regular link. For example, the motif scanning program FIMO can make use of positional priors and therefore has a parameter called "Positional priors" allowing the user to select a Numeric Dataset. This parameter is output in PSP format, but the FIMO program also requires a second auxiliary file based on the same data which should contain binned priors values. Both of these files must be specified on the command line. By using a hidden parameter called "Binned priors" which softlinks to the "Positional priors" parameter, a second file in a different format can thus be created from the same dataset that the user selected for the "Positional priors" parameter.
Conditions (MotifLab v2.0+)
Sometimes a program can have parameters that are only applicable under certain circumstances, which often depends on the settings of other parameters. For example, if the user has selected a value for an optional parameter, a second parameter might have to be specified also, but this second parameter is not required if the first parameter is unspecified. Hence, for the sake of displaying a tidy user-interface dialog for the program, this second parameter should only be shown to the user after a value has been selected for the first parameter. Such context-specific responses to selections in the dialog can achieved be with conditions. A condition is a child-element of a parameter which is set to monitor a parameter and perform certain actions when the value of this parameter is updated. These actions could include showing or hiding other parameters or setting the value of other parameters.Example:
Below is an example with an optional parameter called "Background" which has an associated condition monitoring it. When the user selects a value for this parameter, the condition checks if this value is specified (a background model has been selected) or not (the value is left blank). If a background model was selected, a second parameter called "Other" will be shown in the dialog, if not the "Other" parameter will be hidden.
<parameter type="regular" name="Background" class="BackgroundModel" required="no" >
<condition if="selected" then="Other:show" else="Other:hide" />
</parameter>
|
Each condition must have an if-attribute which specifies a condition that must be met in order to perform an action. (Alternatively, an ifNot-attribute can be used instead to specify that the action should be performed if the condition is not satisfied). If the if-condition is met (or the ifNot-condition is not met), the then-attribute specifies the action to perform. An optional else-attribute can be used to specify an action that should be performed instead of the then-action if the if-condition is not met.
If-attribute:
The if-attribute can have one of the following values
- selected
- value=<allowed values>
- type=<allowed types>
- updated
For Boolean parameters this condition is met if the value is TRUE and not FALSE.
If the condition is based on the "value" of the parameter, the condition will be met if this value equals one of the listed values (multiple values can be separated with vertical bars, e.g. "value=1|2|3"). Note that the value that is used is the value of the parameter as it appears in the GUI dialog and not the value of any selected data object. Hence, if the user has selected a Numeric Variable called "X" (with a value of 54) for the parameter, the value that is checked is "X" and not 54. This condition is thus mostly useful for checking the value of String-type parameters.
If the condition is based on the "type" of the parameter, the condition will be met if the data type of the selected value equals one of the listed types (multiple types can be separated with vertical bars). This could, for example, be used to check if the value for an Integer-type parameter was specified with a literal integer ("type=Integer") or with a Numeric Variable ("type=NumericVariable")
The "updated" condition is always met as long as the user has made selections or updates for this parameter in the dialog (even if the selected value is the same as before).
The condition of the if-attribute will usually refer to the value of the enclosing parameter. However, it is possible to specify that the condition should monitor a different parameter instead by specifying the optional monitor="<parameterName>"-attribute (see example below).
Then- and Else-attributes:
These attributes specify an action to perform when the if-condition is met or not met respectively. Recognized values are:
- show
- hide
- setValue=<somevalue>
- setToValueOf=<parameter>
The specified action will normally be applied to the enclosing parameter, but it is possible to apply the action to a different parameter instead by prefixing the action with the name of that parameter followed by a colon (e.g. "OtherParameter:hide" or "OtherParameter:setValue=7").
Note that it is only possible to specify a single action to perform when the condition is met (or not). However, if it is desirable to perform several actions one can always include multiple conditions for a parameter.
Example 2:
This example is equivalent to the example above and shows an alternative way to accomplish the same effect from a different perspective. In the above example, the condition was associated with the "Background" parameter which monitored itself. Depending on the value of this parameter the actions to be performed, as specified by the then- and else-attributes, were applied to a second parameter named "Other" by prefixing the value of the then- and else-attributes with "Other:". In the example below, the condition is instead associated with the "Other" parameter, but the condition is set to monitor the value of the "Background" parameter by setting the monitor="Background" attribute of the condition. Since the actions to be performed when the condition is met (or not) is to be applied to the enclosing parameter ("Other"), the prefix was dropped from the then- and else-attributes.
<parameter type="regular" name="Other" class="..." required="no" >
<condition monitor="Background" if="selected" then="show" else="hide" />
</parameter>
|
Reports (MotifLab v2.0+)
Often a program that writes its regular results to files will output additional information during execution to either STDOUT or STDERR (or both) to inform the user of the program's progress and report on any errors that have been encountered. The standard way to handle such output by MotifLab is to display each line in the GUI's status bar at the bottom of the screen. Version 2.0 of MotifLab, however, introduced the <report> element which can recognize specific expressions and display them either in the status bar, the log panel or an error dialog. If the program outputs information about how far it has come in its execution in the form of a percentage number or ratio, this information can also be captured and used to set the progress bar in the GUI.
<reports>
<report expression="" target="status" />
<report expression="WARNING:.+" target="log" />
<report expression=".*?next.*" target="log" />
<report expression="ERROR:.+" target="error" />
<report expression=".+?:(\d+)%.*" target="progress" />
</reports>
|
Each <report> element has one required expression argument and two optional arguments target and output. The expression argument specifies a regular expression that MotifLab should look for in the output. If a line sent to STDOUT or STDERR by the program matches a specified expression, that line will be sent to the designated target which can be either "status" (line is displayed in the status bar), "log" (line is displayed in the the log-panel), "error" (line is displayed in an error dialog and the execution of the program is stopped) or "progress". The "progress" target has some special requirements on the regular expression, namely that it must include either one or two capture groups, i.e. expressions enclosed in parentheses that match a number, such as e.g. "(\d+)" in the last example above. If only one capture group is specified, this should match a (percentage) number between 0 and 100 which will be used directly to set the progress in the progressbar. If two such capture groups are specified, the first group should capture a number reporting how many subtasks that have been completed so far and the second group should capture a number reporting the total number of such subtasks (e.g. "processing sequence 23 of 60"). The ratio between the first and second number will then be used to set the progressbar. Note that the specified regular expression must match a whole line in the output by the program and not just a substring. This means that it could be wise to start the expression with ".*?" and end it with ".*" to be sure that the whole line is matched. An empty expression is considered as a wildcard and will match any output. Hence, in the example above, the first report line will display every line of output produced by the program in the status bar, lines starting with the word "WARNING:" or containing the word "next" will be displayed in the log-panel (note that it is possible to specify multiple reports for the same target), and if the program ever outputs a line starting with "ERROR:", MotifLab will end the execution of the program and report this line in an error dialog. The last report statement will search for lines containing any text followed by a colon and an integer number suffixed by a % sign. This integer number will then be used to set the value of the progressbar.
Normally, the line that is matched by the given expression will be displayed to the user. However, it is also possible to state that a different text should be displayed with an optional output argument. For example, the statement "
Cleaning up
If a program creates any additional files or directories during its execution (besides the temporary files created to pass complex parameters), it is prudent to specify these so that MotifLab can perform the necessary clean up after the execution has finished. The <temporary> element is used to specify the names of these temporary files (or directories). The special strings %WORKDIR and %APPDIR explained above can prefix the filenames if necessary.
<program>
...
...
...
<temporary filename="tempfile1" />
<temporary filename="%WORKDIR/tempfile2" />
</program>
|
Configuring data tracks and sources
Datatracks XML configuration file
Documentation is in preparation...Data Formats
Data formats define ways to formally describe the information contained in a data track or other data object and thus allows this information to be written to files and shared between computational tools. MotifLab supports many of the standard bioinformatics data formats that are relevant to regulatory sequence analysis, including e.g. FASTA, GFF and BED for feature data tracks and TRANSFAC or JASPAR formats for describing motif models. Data objects can be output to a selected data format with the output operation. This operation will create a textual representation of the data according to the specified format and store this text in special Output Data objects (shown as separate tabbed panels in MotifLab).The contents of such Output Data objects can then be saved to file. Most data formats can be used for both output and input, meaning that information that has been exported in a specific format can be read back by MotifLab at a later time and used to reconstruct the original data objects. However, a few data formats can only be used for either input or output. For example, MotifLab is able to import data from the compressed binary formats BigBED, BigWIG and 2bit, but is currently not capable of exporting data in these formats. Conversely, information about sequences or motifs can be presented in aesthetically pleasing tables in various HTML-based formats, but MotifLab can not parse this information back again to reconstruct the original sequences or motifs.
Complete versus lossy data formats
Data objects usually have a set of recognized standard properties depending on their type. For example, all sequence objects have a genomic location and strand orientation and motifs have names and PWM models (or IUPAC models). All standard data formats that apply to sequences thus have ways to represent the location and strand of a sequence, and data formats used to describe motifs include descriptions of the name and PWM model. However, in addition to such standard properties, data objects in MotifLab often have non-standard or user-defined properties that are not necessarily supported by standard data formats. Hence, if a data object that contains non-standard properties is exported in a standard data format, these non-standard properties will usually be ignored in the output. Consequently, it will not be possible for MotifLab to fully reconstruct the original data object when reading the information back again with such a data format. Below, we use the term complete when referring to data formats that always support the full set of both standard and non-standard properties, and thus allow data objects to be completely reconstructed from files. Users will never risk loosing information if these formats are used. Conversely, lossy data formats do not save all the necessary information required to fully reconstruct the original data object, and these data formats should then be used with some caution. Potentially complete data formats do not save all the information by default, but can be considered complete if necessary precautions are taken.Below is an incomplete classification of some of the data formats supported by MotifLab
| Data Type | Complete | Potentially complete | Lossy |
|---|---|---|---|
| DNA Datasets | FASTA | ||
| Numeric Datasets | PRIORITY | PSP2, WIG3, BedGraph3 | |
| Region Datasets | GFF4, BED1,5, EvidenceGFF1,5, Region_Properties1 | GTF | |
| Sequences | Location7 | Sequence_Properties1, BED1, Properties1 | |
| Motifs | MotifLabMotif | Motif_Properties1, Properties1 | TRANSFAC, Jaspar, MEME_Minimal_Motif, INCLUSive_Motif_Model, RawPSSM, XMS, HTML_MotifTable, HTML_Matrix, BindingSequences |
| Modules | MotifLabModule | Module_Properties1, Properties1 | |
| Collections | All applicable formats | ||
| Partitions | All applicable formats | ||
| Maps | All applicable formats | ||
| Background models | All applicable formats6 |
- These formats can specify which properties that should be included. Hence, in order to make them complete, all properties must be specified.
- The PSP format can be considered complete only if the "motif width" parameter is set to 0.
- MotifLab allows sequences to overlap with other sequences but still be treated as completely separate with respect to the contents of associated feature tracks. For example, if you have two separate sequences A and B that have the exact same genomic location and add e.g. a conservation track, the conservation track will initially be the same for the two sequences. Later, however, the conservation track can be manipulated with operations or edited with the draw tool so that the track has different contents for sequence A and B. Whereas the data formats PRIORITY and PSP will save the track information from a sequence-centric perspective (representing the information as a list of values for each sequence without any consideration to where the sequence is located), the WIG and BedGraph data formats take on a genome-centric perspective and make a note of the genomic position that each value in the track is associated with (without considering which sequence it belongs to). Hence, when importing back information stored in WIG or BedGraph formats, information pertaining to one sequence can overwrite another sequence if they overlap. However, if none of the sequences overlap with each other, these formats can also be considered complete.
- GFF is only complete for module tracks if the "include module motifs" parameter is selected. GFF currently does not support tracks with other linked regions.
- EvidenceGFF and BED formats are not complete when used with module tracks or linked-region tracks
- The "INCLUSive_Background_Model" is the only background model data format that fully supports meta-data, but such meta-data is not fully supported by MotifLab.
- Provided that the full "10-field" format or a complete custom-format is used
Default data formats
All data types have an associated default format which is the format used for that data type when no other is specificed (e.g. when executing the command "output DataObject" without a following "in format XXX" argument). The default format is also used when importing data objects specified with data injection.| Data type | Default format |
|---|---|
| DNA Datasets | FASTA |
| Numeric Datasets | PRIORITY |
| Region Dataset | GFF |
| Sequences and Sequence Collections | Plain |
| Motifs and Motif Collections | MotifLabMotif |
| Modules and Module Collections | MotifLabModule |
| Partitions | Plain |
| Maps | MapFormat |
| Background models | INCLUSive_Background_Model |
| Expression Profiles | ExpressionProfile |
Feature Dataset formats
FASTA
| Applies to: | DNA Sequence Dataset |
Example of sequence data in FASTA format:
GTAGTCGCTGCACAGTCTGTCTCTTCGCCGGTTCCCGGCC
CCGTGGATCCTACTTCTCTGTCGCCCGCGGTTCGCCGCCC
>ENSG00000100345
GCAGATCACCGCGGTTCCTGGGCAGGGCACGGAAGGCTAA
GCAAGGCTGACCTGCTGCAGCTCCCGCCTCGTGCGCTCGC
>ENSG00000107796
AACACCACCCAGTGTGGAGCAGCCCAGCCAAGCACTGTCA
GGGTAAGTGGCGCCAGGCCAAGGATGTGACTTATAGATTC
The header can contain other information in addition to the name of the sequence if the fields are separated by vertical bars. The fields are in order: sequence name, sequence location, strand orientation and organism/genome build. MotifLab version 2.0 can also recognize a fifth field specifying the gene name and location (position of TSS and TES). All the extra fields are optional, but the order is important, so if you want to include information about the strand, you must also include the sequence location field preceeding it.
Example:
>ENSG00000035403|chr10:75425878-75428077|Direct strand|9606:hg18|VCL:75427878-75549916
The sequence name must not contain spaces or characters other than letters, numbers or underscores. If the name contains spaces, only the first part of the name will be used. If the name contains other illegal characters, an error will be reported. The location must be given as "chromosome:start-end" (where the "chr" prefix for the chromosome is optional). For the orientation, strings starting with "direct", "+" or "1" are interpreted as the direct strand whereas strings starting with "reverse" or "–" are interpreted as the reverse strand (other strings will just default to direct strand). The "organism/genome build" field should be specified as two values separated by a colon, where the first value is an integer taxonomy identifier (or known organism name) and the second value is the genome build. Optionally, the genome build can be stated alone and the system will then try to infer the organism. The fifth "gene location" field introduced in MotifLab v2.0 is on the form "gene name:TSS-TES".
Arguments
| Name | Description |
|---|---|
| Strand orientation | This parameter controls which strand to output for each sequence. Valid options are "Direct" (output sequence from genomic direct strand), "Reverse" (output sequence from genomic reverse strand) and "Relative" (output sequence data relative to the orientation of the sequence. I.e. use same strand as the strand the sequence originates from). |
| Header | Specifies what information to include in the header (after the > sign). The default is to output only the name of the sequence, but additional fields (separated by vertical bars) can also be output, such as the genomic location of the sequence, the strand orientation of the sequence and the genomic build of the sequence. |
| Column width | The number of sequence bases to output on each line. If the length of the sequence is longer than the specified column width, the sequence data will be split across multiple lines. A common value is 80, but the special value of 0 can be used to specify that the whole sequence should be output on one single line. |
| Extra space | If selected, an extra empty line will be added after the sequence data for each sequence (and before the header of the next sequence) to separate the sequences visually. Note that some external programs might not be able to parse FASTA files correctly if extra lines are added. |
See also: output, DNA Sequence Dataset
2bit
| Applies to: | DNA Sequence Dataset |
Arguments
| Name | Description |
|---|---|
| Keep masks |
If selected, lowercase letters in the DNA sequence will be kept as is ("masked").
If not selected, all bases will be in uppercase.
NOTE: The current implementation of the 2bit format in MotifLab is very inefficient when this option is selected, so it is not recommended to use it. |
See also: output, FASTA, DNA Sequence Dataset
WIG
| Applies to: | Numeric Dataset |
A WIG file consists of one or more blocks where each block starts with a declaration line and is followed by lines defining data elements. There are two main formatting options: fixedStep and variableStep, and each block can have different formatting as described in the block's declaration line. Note that while MotifLab is capable of reading blocks in both of these formats, it will only produce output in variableStep format (with span=1).
variableStep
variableStep format is designed for data with irregular intervals between data points, and is the more commonly used format. It begins with a declaration line, followed by two columns containing chromosome positions and data values.
The declaration line begins with the word "variableStep" and is followed by space-separated key-value pairs:
- chrom (required) - name of chromosome
- span (optional, defaults to 1) - the number of bases that each data value should cover
Without span:
300701 12.5
300702 12.5
300703 12.5
300704 12.5
300705 12.5
With span:
300701 12.5
Both of these examples will display a value of 12.5 at position 300701-300705 on chromosome 2.
fixedStep
fixedStep format is designed for data with regular intervals between data points and is the more compact of the two wiggle formats. It begins with a declaration line, followed by a single column of data values.
The declaration line begins with the word "fixedStep" and is followed by space-separated key-value pairs:
- chrom (required) - name of chromosome
- start (required) - start point for the data values
- step (required) - distance between data values
- span (optional, defaults to 1) - the number of bases that each data value should cover
11
22
33
Displays the values 11, 22, 33 as single-base features, on chromosome 3 at positions 400601, 400701 and 400801 respectively.
With span:
11
22
33
Displays the values 11, 22, 33 as 5-base features, on chromosome 3 at positions 400601-400605, 400701-400705 and 400801-400805 respectively.
Data values
Wiggle element data values can be integer or real, positive or negative. Chromosome positions are 1-relative, i.e. the first base is 1. Only positions specified have data; unspecified positions will be empty.
See also: output, Numeric Dataset
BigWig
| Applies to: | Numeric Dataset |
See also: output, WIG, Numeric Dataset
BedGraph
| Applies to: | Numeric Dataset |
Each line in BedGraph format contains four columns where the first three define a chromosomal region (similar to the first three columns of the BED format) and the last column specifies a numeric value that applies to all the positions within that region.
Example:
chr19 49302300 49302600 -0.75
chr19 49302600 49302900 -0.50
chr19 49302900 49303200 -0.25
chr19 49303200 49303500 0.0
chr19 49303500 49303800 0.25
chr19 49303800 49304100 0.50
Arguments
| Name | Description |
|---|---|
| Add CHR prefix | If selected, the prefix "chr" will be added before the chromosome number (e.g. chromosome 12 will be output as "chr12" rather than just "12"). |
| Coordinate system | Selects whether the coordinates in the are in the standard BED-coordinate system (with the chromosome starting at position 0 and end-coordinates being exclusive) or in the format used by e.g. GFF, where the chromosome starts at position 1 and both start- and end-coordinates are inclusive. |
See also: output, WIG, BED, Numeric Dataset
PRIORITY
| Applies to: | Numeric Dataset |
Example:
0.118,0.188,0.839,0.887,0.91,0.898,0.903,0.873,0.0,0.002,0.003,0.001,0.0,0.994,0.996
>ENSG00000100345
0.998,0.999,0.998,0.997,0.997,0.998,0.998,0.982,0.994,1.0,1.0,1.0,1.0,1.0,1.0
>ENSG00000107796
0.444,0.519,0.999,1.0,1.0,1.0,1.0,1.0,1.0,1.0,0.992,0.997,0.994,0.975,0.396
Arguments
| Name | Description |
|---|---|
| Orientation |
This parameter dictates the order in which to list the values for each
sequence. The default setting is "Relative" and the valid settings are:
|
| Separator | The separator to use between the data values. The default is "Comma", but other valid options are "Space" and "TAB". |
See also: PSP, FASTA, output, Numeric Dataset
PSP
| Applies to: | Numeric Dataset |
Example:
0.118 0.188 0.839 0.887 0.91 0.898 0.903 0.873 0.0 0.002 0.003 0.001 0.0 0.0 0.0
>ENSG00000100345 4
0.998 0.999 0.998 0.997 0.997 0.998 0.998 0.982 0.994 1.0 1.0 1.0 0.0 0.0 0.0
>ENSG00000107796 4
0.444 0.519 0.999 1.0 1.0 1.0 1.0 1.0 1.0 1.0 0.992 0.997 0.0 0.0 0.0
Arguments
| Name | Description |
|---|---|
| Orientation |
This parameter dictates the order in which to list the values for each
sequence. The default setting is "Relative" and the valid settings are:
|
| Motif width | This value (hereafter called W) will be output after the sequence name in the header for each sequence, and the last W-1 values for each sequence will be set to 0.0 (as required by the PSP format). |
| Include width | If selected (default), the motif width parameter will be output after the sequence name in the header. Note that the W-1 values for each sequence will still be set to 0.0 even if the motif width is not included in the header. |
| Normalize | The original PSP specification requires that all the values lie between 0 and 1 and that the sum of values for each sequence is no greater than 1.0. The "Normalize" parameter can be used to normalize all the values so that the output conforms to these requirements. The default setting is to not perform any normalization, but it is also possible to normalize the values by dividing each position with the largest value for each sequence ("Max 1") or by dividing the value in each position with the sum across all positions ("Sum to 1"). |
See also: PRIORITY, FASTA, output, normalize, Numeric Dataset
GFF
| Applies to: | Region Dataset |
The fields are in order:
- The name of the sequence
- The source of the feature
- The feature type
- The start coordinate of the region
- The end coordinate of the region
- A score value for the region
- The orientation of the region. This can be "+" or "-" (or "." if orientation is unspecified)
- The reading frame. The value of this field is either 0, 1 or 2 (or "." if the frame does not apply)
- Additional attributes. This optional field consists of a list of attributes separated by semicolon. Each attribute has a key (or "tag") followed by value for the attribute (separated by an equals sign).
NOTE:
When importing regions from a GFF-file, the sequence name in the first column must correspond to the name of an existing sequence in MotifLab, and the region will then be added to that sequence. If the first column contains a chromosome name, it will only be added to a sequence if there is a sequence that is actually named after the chromosome; it is not enough that the sequence covers the chromosomal segment that the region from the GFF-file falls within. When the first column contains chromosome names, it is suggested instead to use the GTF format (or convert the file to BED format).
Sequences output in GFF format are output according to the currently selected sorting order of the sequences, but within each sequence the user can specify whether to sort the regions by position, score or type. The start and end positions of each region (fields 4 and 5) can be output as either genomic coordinates or as positions relative to the start of the sequence by setting the "Position" option to either "Genomic" or "Relative". If the "Relative" setting is chosen, the "Relative-offset" and "Orientation" settings will also apply. The "Relative-offset" setting specifies the coordinate of the first position in the sequence. This will normally be 1 but can be set to other values if needed (for instance 0). The "Orientation" setting specifies which orientation to use to determine the relative region coordinates. For example, if a 100 bp long sequence on the direct strand has a binding site region from position 80 to 90, the start and end coordinates will be [80,90] if the "Direct" strand orientation is selected or [10,20] if the "Reverse" orientation is selected. If the "Orientation" is set to "From Sequence" the strand orientation will be selected based on the orientation of the sequence itself, so that sequences on the direct strand will be output in direct orientation and those on the reverse strand will be output in reverse orientation. If the "Opposite" strand orientation is selected, the orientation will be the opposite of the orientation of the sequence.
If the standard GFF format is not adequate, the "Format" setting can be used to specify an alternative output format. The alternative format is specified by a string consisting of a mix of literal characters and special field codes surrounded by braces (e.g. {START} ). For each region, the field codes in the format string (if recognized) will be replaced by the corresponding value of the field as it applies to the target region before the string is output. Some recognized fields are: SEQUENCENAME, FEATURE, SOURCE, START, END, SCORE, STRAND and TYPE (note the capitalization). TABs can be represented with the escape character \t.
For example, the following output format:
will produce output that looks like this
Binding site for M00253 at 3-10 with score=3.801 in sequence ENSG00000116741
Binding site for M00313 at 8-15 with score=5.697 in sequence ENSG00000116741
Arguments
| Name | Description |
|---|---|
| Position | Specifies whether the coordinate positions [start-end] for a region should be given relative to the start of the chromosome ("Genomic"), relative to the upstream start of the sequence ("Relative") or relative to the transcription start site associated with the sequence ("TSS-Relative") |
| Relative-offset | If the "Position" setting is set to "Relative", this offset-value specifies what position the first base in the sequence should start at (common choices are 0 or 1). If the "Position" setting is "TSS-Relative", a value of 0 here will place the TSS at position +0 whereas any other value will place the TSS at position +1 (and the coordinate-system will then skip 0 and go directly from -1 to +1) |
| Orientation | Orientation of relative coordinates (only applicable if the "Position" setting is "Relative"). |
| Sort1,Sort2,Sort3 | These three parameters control how to sort the regions in the output. The regions will be sorted first by "Sort1", then by "Sort2" (for regions with similar values for the "Sort1" property) and finally by Sort3. Valid choices for these parameters are "Position", "Type" and "Score" and the three parameters should preferably have different values. The default choice is to first so by "Position", then "Type" and finally "Score". |
| Include module motifs | If selected, the constituent single TF binding sites making up a cis-regulatory module will also be included for each module region. Hence, if a module consists of three TFBS, the module region will be output first on one line followed by three lines containing each of the TFBS regions. The third column in the output will have the value "module" for the module regions and "motif" for the individual TFBS. Also, a "module_identifier" is output on each line that can be used to group together a module entry with its corresponding motif (TFBS) entries. |
| Skip header lines | This (hidden) parameter can be used to specify a number of lines that should be skipped at the start of the file (default is 0). These lines are suspected to contain comments or other information that do not conform to standard GFF format and would therefore result in parsing errors if treated as regular input. |
| Format |
The "Format" parameter allows you to specify a
different format to use rather than the standard GFF fields. In
additional to literal text, the format string can
contain field-codes surrounded by braces,
e.g. {TYPE}. These field codes will be replaced by the
corresponding property value of the region. Standard recognized
field codes include: SEQUENCENAME,START,END,TYPE,SCORE,STRAND and
ATTRIBUTES. Other field codes can be used to refer to user-defined
properties. Tabs can be inserted using \t and extra newlines
can be inserted with \n.
Example: Use the following format string to output a comma-separated list with the type of the region plus start and end coordinates in the sequence: {TYPE},{START},{END} |
See also: output, EvidenceGFF, Region Dataset
GTF
| Applies to: | Region Dataset |
See also: output, GFF, Region Dataset
EvidenceGFF
| Applies to: | Region Dataset |
The additional properties to output are specified as a string in the "Evidence" setting. This setting should be a list of comma-separated fields in "key=value" format. (Alternatively, the list can be separated by semicolons instead of commas and colons can be used instead of "=" to separate the name of the key from its value).
The "key" can either refer to a known feature dataset or be one of the special keywords region, motif, module, sequence or text.
The proper format of the "value" will depend on the type of the key as described in the table below:
|
If the key is the special keyword "region" the "value" can refer to any property associated with the region.
Some common region properties are:
|
If the key is the special keyword "motif" the following formats for "value" are recognized:
|
|
If the key is the special keyword "module" the "value" can be any standard or user-defined module property. This feature was added in MotifLab v2.0.-3. Note that the "module" keyword is only applicable for module tracks where each region refers to a known cis-regulatory module type. If the region is not associated with a module, the special code "N/A" will be output. |
|
If the key is the special keyword "sequence" the following formats for "value" are recognized: (requires MotifLab version 1.05+)
|
|
If the key is the special keyword "text" then the
corresponding value will be output verbatim. E.g. the evidence code "text=BindingSite" will output "BindingSite" for every region. |
If the key is the name of a DNA Sequence Dataset the following formats for "value" are recognized:
|
If the key is the name of a Numeric Dataset the following formats for "value" are recognized:
|
|
If the key is the name of a Region Dataset (hereafter called the "target dataset") the "value" should be in the following format: <operator> [qualifiers] <condition> [range] [additional] The qualifiers field is optional but can contain a space-separated list of keywords as defined below. The range field is only required when the condition is "within". The additional field can be added when the operator is "list". Allowed values for this field are described below in connection with the list-operator. Based on the condition, range and qualifiers, a set of target regions will be obtained from the target dataset. The following conditions will determine which target regions are included in this set:
After the set of target regions have been obtained based on the condition and filtered based on the selected qualifiers, the choice of operator will determine the final output. EvidenceGFF recognizes the following operators:
Note that if the "target dataset" is the same as the region dataset being currently output in EvidenceGFF format, the current region being output will never be included in the set of target regions described here. Examples: (keys are assumed to be referring to known Region Datasets) DNaseHS=is overlapping Will output YES or NO depending on whether the current region being output overlaps with any regions in the DNaseHS track. ChIP_Seq_tags=count covering Will output the number of ChIP_Seq_tags that are completely covering the current region being output (so that the current region is fully inside the tag region) TFBS=list non-overlapping interacting within 20 with scores and distances Will list the type names of TFBS regions that are overlapping an interval extending 20 bp on either side of the current region but not overlapping with the current region itself. The target regions must be associated with motifs that are known to interact with the motif associated with the current region. The score of the target region will be output in parenthesis after its type name and this will be followed by the distance between the target region and the current region in brackets. |
For example, the following "Evidence" format:
will add 4 new fields to the GFF format. The first new field will contain a short name of the motif associated with the region being output. The second field will contain the average value of the "Conservation" track within the interval spanned by the region. The third field will contain a YES or NO value depending on whether or not the region overlaps with a region in the "Repeats" track, and the fourth and last field will contain a list of type names for regions in the "TFBS" track that are within 30 bp of the current region. The output could look something like this:
RPRM BindingSites M00253 296 303 3.801 + . V$CAP_01 0.235 Yes M00313
RPRM BindingSites M00313 301 308 5.697 + . V$GEN_INI2 0.0 Yes M00253,M00315
...
Arguments
| Name | Description |
|---|---|
| Position | Specifies whether the coordinate positions [start-end] for a region should be given relative to the start of the chromosome ("Genomic"), relative to the upstream start of the sequence ("Relative") or relative to the transcription start site associated with the sequence ("TSS-Relative") |
| Relative-offset | If the "Position" setting is set to "Relative", this offset-value specifies what position the first base in the sequence should start at (common choices are 0 or 1). If the "Position" setting is "TSS-Relative", a value of 0 here will place the TSS at position +0 whereas any other value will place the TSS at position +1 (and the coordinate-system will then skip 0 and go directly from -1 to +1) |
| Orientation | Orientation of relative coordinates (only applicable if the "Position" setting is "Relative"). |
| Sort1,Sort2,Sort3 | These three parameters control how to sort the regions in the output. The regions will be sorted first by "Sort1", then by "Sort2" (for regions with similar values for the "Sort1" property) and finally by Sort3. Valid choices for these parameters are "Position", "Type" and "Score" and the three parameters should preferably have different values. The default choice is to first so by "Position", then "Type" and finally "Score". |
| Include header | If selected, a single header line (starting with #) will be output at the beginning of the output-document. The header contains a specification of all the fields included in the output. |
| Skip standard fields | If selected, the standard GFF fields will not be output, only the evidence fields. |
| Boolean format | This parameter specifies how boolean values should be formatted in the output. Either as "Yes" versus "No" (alternatively "Y" versus "N"), "True" versus "False" (alternatively "T" versus "F") or "1" versus "0". |
| Evidence format | Specifies how the "evidence" should be output for each region. Options are to output each evidence value in a column of its own or to output all evidences in a single column in key=value pairs (separated by semicolons). |
| Evidence | The "evidence" parameter should be a comma-separated list of key=value pairs specifying additional information that should be output for each region. See above for a complete description of recognized evidence codes. |
See also: output, GFF, Region Dataset
BED
| Applies to: | Region Dataset |
- Chromosome: The name of the chromosome or scaffold.
(Chromosome names can be given with or without the "chr" prefix) - Chromosome start: Start position of the region in chromosomal coordinates.
- Chromosome end: End position of the region in chromosomal coordinates.
- Name: The type name of the region.
- Score: The score for the region.
- Strand: The strand orientation of the region.
This can be either "+" (direct strand) or "-" (reverse strand), or even "." if the orientation is undetermined.
Example:
chr10 75427002 75427007 M00028 4.686097666202365 +
chr10 75427002 75427007 M00029 4.486802949517 +
chr10 75427003 75427014 M00472 8.447923342601406 -
chr17 8474690 8474701 M00073 7.7850394311299675 +
chr17 8474710 8474718 M00428 6.149151076269675 +
chr17 8474719 8474730 M00507 8.998892822877837 -
Arguments
| Name | Description |
|---|---|
| Add CHR prefix | If selected, the prefix "chr" will be added before the chromosome number (e.g. chromosome 12 will be output as "chr12" rather than just "12"). |
| Start position | Selects the position of the first base in a chromosome. The default is 0 for BED, but it is usually 1 for other data formats so MotifLab allows the start position to be specified as either 0 or 1. |
| Coordinate system | Selects whether the coordinates in the BED-file are in the standard BED-coordinate system (with the chromosome starting at position 0 and end-coordinates being exclusive) or in the format used by e.g. GFF, where the chromosome starts at position 1 and both start- and end-coordinates are inclusive. This parameter replaces the "Start position" parameter that was used in earlier MotifLab versions. |
| Format | This optional parameter can be used to explicitly define the contents of each line in the BED-file if a non-standard format is used. The format should be defined as a comma-separated list of column names. The default format assumes the BED-file contains the following six columns: "CHROMOSOME, START, END, TYPE, SCORE, STRAND". A line is allowed to contain fewer columns than the format specifies, in which case the missing columns are simply ignored. If the file contains additional columns that the user wants to import, the names of these columns must be included in the format specification. For example, if the file has an additional column containing the property "GeneID" after the STRAND column, the format parameter should be set to "CHROMOSOME, START, END, TYPE, SCORE, STRAND, GeneID". It is also possible to skip columns by replacing the column name with an asterisk (*). For example, if a non-standard BED-file contains the columns "CHROMOSOME, START, END, TYPE, SCORE, GeneID", and a user wants to import all of these properties except for the SCORE property, the format parameter can be set to "CHROMOSOME, START, END, TYPE, *, GeneID" (with SCORE replaced by * in the format definition). |
See also: output, Region Dataset
BigBed
| Applies to: | Region Dataset |
Arguments
| Name | Description |
|---|---|
| Custom fields | This optional parameter can be used to declare additional fields in the BigBed file. A BigBed file is required to contain at least three fields which are in order: CHROMOSOME, START and END. If a line contains more than three fields, the next fields must be TYPE, SCORE and STRAND (+/-/.). If a line contains more than these six fields, the rest will be regarded as custom fields. MotifLab can read these custom fields and add their values to the region as user-defined properties, but to do that the fields must be identified by supplying a comma-separated list of property names. For example, if the Custom fields parameter is set to "count,gene", each entry in the BigBed file is expected to have (at least) 8 fields where the 7th field is named "count" and the 8th field is named "gene". If the name of a custom field is set to "*" it will be ignored. Thus, if the Custom fields parameter is set to "*,gene", the value in the 7th field will be ignored but the value in the 8th field will be added to the region as a user-defined property named "gene". |
See also: output, BED, Region Dataset
Region_Properties
| Applies to: | Region Dataset |
Example of regions output (one on each line) with the Sequence format string "{sequencename}" and Region format "{type} ({motif:short name})\t{sequence:chromosome string}:{genomic start}-{genomic end} [{orientation string}] => {sequence}"
ENSG00000035403 M00048 (F$ADR1_01) chr10:75427746-75427751 [Direct] => TGGGGC
ENSG00000035403 M00028 (I$HSF_01) chr10:75427761-75427765 [Direct] => CGAAA
ENSG00000100345 M00344 (P$RAV1_02) chr22:35113793-35113804 [Direct] => CTCACCTGAACC
ENSG00000100345 M00428 (V$E2F1_Q3) chr22:35113815-35113822 [Reverse] => GTTCCCGG
ENSG00000100345 M00497 (V$STAT3_02) chr22:35113817-35113824 [Reverse] => CTGTTCCC
ENSG00000100345 M00029 (F$HSF_01) chr22:35113818-35113822 [Direct] => GGAAC
ENSG00000173531 M00482 (V$PITX2_Q2) chr3:49701607-49701617 [Direct] => TGTCATCCCAG
ENSG00000173531 M00500 (V$STAT6_02) chr3:49701617-49701624 [Reverse] => ACCTTCCC
ENSG00000173531 M00048 (F$ADR1_01) chr3:49701652-49701657 [Direct] => AGGGGT
ENSG00000173531 M00378 (V$PAX4_03) chr3:49701653-49701664 [Reverse] => TACCTCCACCCC
ENSG00000173531 M00048 (F$ADR1_01) chr3:49701657-49701662 [Direct] => TGGAGG
Arguments
| Name | Description |
|---|---|
| Layout | This parameter controls the general layout of the output. The two available choices are "one sequence per line" (which will output selected information about the sequence followed by selected information about every region within that sequence) and "one region per line" (which will output information on only one region per line, preceeded by information about its parent sequence). |
| Sequence format |
This parameter specifies the information to output for each sequence. In additional to literal text, the format string can contain property codes surrounded by braces,
e.g. {CHROMOSOME}. These property codes will be replaced by the corresponding property values of the sequence in the output. Some standard property codes include SEQUENCENAME, START, END and STRAND. See the documentation for the "Sequence_Properties" data format for a comprehensive list of standard sequence properties. Note that names of standard properties are case-insensitive but the names of user-defined properties are case-sensitive. Use the escape character \t to insert a tab and \n to insert a line break. If you leave the field empty it will take on the default value, but you can set it to * (single asterisk) to signal that the field should not be output at all. Note that leading or trailing whitespace in the format string will be ignored, but you can use the escape character \s to represent spaces instead. Tip: If you don't want to output sequence properties at the beginning of the line but rather mix these in between the other region properties, set the sequence format to * (empty) and use property codes prefixed with "sequence:" in the Region format parameter. |
| Region format |
This parameter specifies the information to output for each region.
In additional to literal text, the format string can contain property codes surrounded by braces as explained for the Sequence format parameter above.
Standard region properties include:
Use the escape character \t to insert a tab and \n to insert a line break in the format string. If you leave the field empty it will take on the default value, but you can set it to * (single asterisk) to signal that the field should not be output at all. Note that leading or trailing whitespace in the format string will be ignored, but you can use the escape character \s to represent spaces instead. A note on coordinate systems: The "start" and "end" properties will output the start (and end) position of the region relative to the start of the parent sequence on the direct strand. The relative start and end properties outputs these positions relative to the beginning of the parent sequence, which in this case will be strand dependent. The genomic start and end properties outputs positions relative to the start of the chromosome (as long as the genomic location of the parent sequence is known). The TSS/TES-relative properties will output the start and end relative to the position of the TSS/TES of the gene associated with the parent sequence. All of these coordinate systems start at position 0 except for genomic coordinates which start at 1. If you want these positions to start at 1 instead (or 0 for genomic coordinates) you can explicitly add [0] or [1] after the property name, e.g.: {relative start[1]}, {genomic start[0]} or {TSS-relative start[1]}. 1-indexed coordinates relative to TSS/TES work a little bit different than regular coordinate systems since the TSS/TES will be placed at +1 but the immediate upstream position will be called -1. This, in effect, will skip the 0-position: ..., -3, -2, -1, +1 [TSS], +2, +3, ... This choice will affect the positive coordinates of regions downstream of TSS/TES (or regions spanning TSS/TES) but not the negative coordinates of upstream regions. |
| Sequence delimiter | The delimiter text that separates the sequence information from the region information. The default is a TAB (\t). If you leave the field empty it will take on the default value, but you can set it to * (single asterisk) to signal that the delimiter should be empty. Note that leading or trailing whitespace in the string will be ignored, but you can use the escape character \s to represent spaces instead. |
| Region delimiter | The delimiter text that separates the information of different regions. This only applies when multiple regions are output to the same line. The default is a TAB (\t). If you leave the field empty it will take on the default value, but you can set it to * (single asterisk) to signal that the delimiter should be empty. Note that leading or trailing whitespace in the string will be ignored, but you can use the escape character \s to represent spaces instead. |
See also: output, Sequence_Properties, Motif_Properties, Module_Properties
Motif formats (and module formats)
MotifLabMotif
| Applies to: | Motif and MotifCollection |
Example:
#
#ID = M00002
#Short = V$E47_01
#Long = E47 (E2A immunoglobulin enhancer binding factor)
#W = 15
#Class = 1.2.1.0
#Factors = E47
#Organisms = human (Homo sapiens)
#Interactions = M00001,M00002,M00058,M00065,M00066,M00068,MA0048,MA0081,M00454,MA0092
#Alternatives = M00065,M00066,M00071,M00222,MA0091
#Transfac class = C0010
4.0 4.0 3.0 0.0
2.0 5.0 4.0 0.0
3.0 2.0 4.0 2.0
2.0 0.0 9.0 0.0
0.0 11.0 0.0 0.0
11.0 0.0 0.0 0.0
0.0 0.0 11.0 0.0
1.0 2.0 8.0 0.0
0.0 0.0 0.0 11.0
0.0 0.0 11.0 0.0
0.0 0.0 4.0 7.0
1.0 4.0 3.0 3.0
1.0 6.0 2.0 2.0
1.0 4.0 4.0 2.0
1.0 4.0 2.0 3.0
#ID = M00001
#Short = V$MYOD_01
#Long = MyoD (myoblast determination gene product)
#W = 12
#Class = 1.2.2.0
#Factors = MyoD,MyoD (376 AA),MyoD (275 AA)
#Organisms = chick (Gallus gallus),rat (Rattus norvegicus),human (Homo sapiens)
#Interactions = M00001,M00002,M00004,M00006,M00222,M00223,M00225,M00231,M00232
#Alternatives = M00184
#Transfac class = C0010
1.0 2.0 2.0 0.0
2.0 1.0 2.0 0.0
3.0 0.0 1.0 1.0
0.0 5.0 0.0 0.0
5.0 0.0 0.0 0.0
0.0 0.0 4.0 1.0
0.0 1.0 4.0 0.0
0.0 0.0 0.0 5.0
0.0 0.0 5.0 0.0
0.0 1.0 2.0 2.0
0.0 2.0 0.0 3.0
1.0 0.0 3.0 1.0
Arguments
| Name | Description |
|---|---|
| Include non-standard fields | If selected, information about non-standard, user-defined motif properties will also be included. |
| Include derived fields | If selected, motif properties that are derived from other properties, such as GC-content, IC-content and IUPAC consensus string (all derived from the matrix representation), will also be included in the output. |
| Include color info | If selected, information about the current colors used for the motifs in MotifLab will also be included in the output. When a file containing color information is imported into motiflab, the motifs will be assigned their specified colors. If an imported motif file does not contain color information, the motifs will be assigned arbitrary colors. |
See also: INCLUSive_Motif_Model, output
MotifLabModule
| Applies to: | Motif and MotifCollection |
Each new module is introduced with the line:
This is followed by a list of the motifs involved in the module:
Note that the "module motif" names in the mentioned list are not single motif identifiers referencing Motif objects, but rather descriptive motif names that are internal to the module (the "module motif" names must be unique within the module). Each such "module motif" can be represented by multiple single motif objects, as described by lines in the following format:
Additional constraints regarding the motifs within the module can also be specified, for example the maximum length of the module:
Whether the motifs in the module must appear in the order they are listed in the "Motifs = " line or if they can appear in any order.
The module motifs might also have specific orientations relative to each other.
Or constraints on the distance between pairs of consecutive motifs in ordered modules.
Example:
#
#ModuleID = MOD0001
Motifs = STAT,GATA
Ordered = false
MaxLength = 200
Motif(STAT) = M00225,M00492,M00493,M00494,M00500,M00496
Motif(GATA) = M00351,M00350,M00076,M00203,M00077,M00075,M00347,M00346
#ModuleID = MOD0002
Motifs = SRY,AP1
Ordered = true
MaxLength = 200
Motif(SRY) = M00160,M00148
Motif(AP1) = M00041,M00172,M00039,M00517,M00040,M00113,M00114,M00174,M00115
Orientation(SRY) = Direct
Orientation(AP1) = Reverse
#ModuleID = MOD0003
Motifs = STAT,ER,MYC
Ordered = true
MaxLength = 200
Motif(STAT) = M00225,M00492,M00493,M00494,M00500,M00496,M00497,M00498
Motif(ER) = M00191
Motif(MYC) = M00055,M00322,M00006,M00005,M00007
Orientation(STAT) = Direct
Orientation(ER) = Reverse
Distance(STAT,ER) = [5,10]
Distance(ER,MYC) = [0,16]
Arguments
| Name | Description |
|---|---|
| Include single motifs | The single motifs listed in the "Motif(x)=..." lines must reference Motif objects that are already known to MotifLab. It is possible to include descriptions for the motifs involved in the modules in a MotifLabModule file, so that the file will contain everything that is needed to restore the modules on import. The motif descriptions will be appended after the module descriptions at the end of the file in MotifLabMotif format. |
| Include module color info | If selected, information about the current colors used for the modules in MotifLab will also be included in the output. When a file containing color information is imported into motiflab, the modules will be assigned their specified colors. If an imported module file does not contain color information, the modules will be assigned arbitrary colors. |
See also: INCLUSive_Motif_Model, output
INCLUSive_Motif_Model
| Applies to: | Motif and Motif Collection |
The file must start with a comment line which identifies the format (#INCLUSive Motif Model v1.0).
Next follows the PWM description of a first motif, starting with some comment lines. The first comment line describes a unique motif identifier (#ID). The second comment line shows a motif score (#Score) which can be a score that is computed from the PWM or any other score that reflects the importance of the motif being described. The following two lines give the PWM length (#W) and a consensus description (#Consensus) of the motif. A consensus description is derived from the information available in the PWM; it is a string-based sequence representation of the motif in IUPAC code symbols (A,C,G,T,n,s,w) that describes the most likely nucleotide(s) on each position in the motif (n = any of A,C,G,T, s = C or G, w = A or T. Note that MotifLab can use additional IUPAC codes as well).
The comment lines are immediately followed by the values that make up the PWM (matrix) : each line describes the tab-separated probabilities (Pr) for nucleotide A, C, G and T on a given position in the motif. The number of lines must equal the length of the motif (#W). The probabilities described in a PWM can be frequencies (normalized values between 0 and 1 and the sum of a row equals 1), or they can be represented as counts (values can be higher than 1 and zeros are also common).
MARK : decimal numbers in a PWM must be described using a DOT (not a comma) e.g. 0.54 (not 0,54).
Pr(A,2) Pr(C,2) Pr(G,3) Pr(T,4)
...
Pr(A,W) Pr(C,W) Pr(G,W) Pr(T,W)
The motif description ends with a blank line return. The second and following motifs are described in exactly the same way, each time separated from each other by a blank line. The end of the file is recognized by the last blank line return. Note that there is no explicit numbering of the motifs in the file.
Example:
#
#ID = M00001
#W = 12
#Consensus = srACAGGTGkyG
1.0 2.0 2.0 0.0
2.0 1.0 2.0 0.0
3.0 0.0 1.0 1.0
0.0 5.0 0.0 0.0
5.0 0.0 0.0 0.0
0.0 0.0 4.0 1.0
0.0 1.0 4.0 0.0
0.0 0.0 0.0 5.0
0.0 0.0 5.0 0.0
0.0 1.0 2.0 2.0
0.0 2.0 0.0 3.0
1.0 0.0 3.0 1.0
#ID = M00002
#W = 10
#Consensus = GGGGCGGGGT
2.0 1.0 6.0 2.0
3.0 1.0 6.0 1.0
0.0 0.0 11.0 0.0
0.0 0.0 11.0 0.0
0.0 8.0 2.0 1.0
3.0 0.0 6.0 2.0
0.0 1.0 7.0 3.0
1.0 0.0 8.0 2.0
1.0 2.0 7.0 1.0
3.0 2.0 0.0 6.0
See also: output
RawPSSM
| Applies to: | Motif and Motif Collection |
Example (in "Horizontal" orientation):
2 3 0 0 0 3 0 1 1 3
1 1 0 0 8 0 1 0 2 2
6 6 11 11 2 6 7 8 7 0
2 1 0 0 1 2 3 2 1 6
>M00002
1 2 3 0 5 0 0 0 0 0 0 1
2 1 0 5 0 0 1 0 0 1 2 0
2 2 1 0 0 4 4 0 5 2 0 3
0 0 1 0 0 1 0 5 0 2 3 1
Arguments
| Name | Description |
|---|---|
| Format | If the "Default" format is selected, the matrix will be output exactly as it is represented in each motif (which can be either a count matrix, a frequency matrix or a log-odds matrix). However, if the "Frequencies" format is selected, all matrices will be converted to frequency matrices before being output. |
| Orientation | The orientation can either be "Vertical" or "Horizontal". If a "Vertical" orientation is selected, the matrix will consist of four columns corresponding to each of the bases A, C, G and T and it will have N rows (where N is the length of the motif). If a "Horizontal" orientation is selected, the matrix will consist of four rows corresponding to each of the bases and the matrix will have N columns (one for each position). |
| Delimiter | Specifices the character used to separate the columns in the matrix. The default delimiter is "Tab", but other choices are "Space", "Comma" and "Semicolon". |
| Header | Specifies which information to include in the header for each motif. The possible options are: include only the motif ID ("ID"), include both the motif ID and motif name (short name) separated with a space ("ID Name") or include both the motif ID and motif name separated with a hyphen ("ID-Name"). |
See also: output
TRANSFAC
| Applies to: | Motif and Motif Collection |
- AC : This field will map to the motif identifier.
- ID : This field will map to the "short name" of the motif.
- NA : This field will output a cleaned up version of the "short name"of the motif (stripped of prefixes and suffixes) but is not used for input.
- DE : This field will map to the "long name" of the motif.
- BF : This field can contain the name of a TF binding to the motif and the organism in which this happens. This field is used for input to populate the "binding factors" and "organisms" properties of the motif, but it is not used for output.
- P0 : This code marks the start of the matrix field
Example:
XX
//
AC M00001
XX
ID V$MYOD_01
XX
NA MYOD
XX
DE MyoD (myoblast determination gene product)
XX
P0 A C G T
01 1 2 2 0 S
02 2 1 2 0 R
03 3 0 1 1 A
04 0 5 0 0 C
05 5 0 0 0 A
06 0 0 4 1 G
07 0 1 4 0 G
08 0 0 0 5 T
09 0 0 5 0 G
10 0 1 2 2 K
11 0 2 0 3 Y
12 1 0 3 1 G
XX
//
AC M00002
XX
ID V$E47_01
XX
NA E47
XX
DE E47 (E2A immunoglobulin enhancer binding factor, also known as Transcription factor 3 (TCF3))
XX
P0 A C G T
01 4 4 3 0 V
02 2 5 4 0 S
03 3 2 4 2 N
04 2 0 9 0 G
05 0 11 0 0 C
06 11 0 0 0 A
07 0 0 11 0 G
08 1 2 8 0 G
09 0 0 0 11 T
10 0 0 11 0 G
11 0 0 4 7 K
12 1 4 3 3 N
13 1 6 2 2 C
14 1 4 4 2 N
15 1 4 2 3 N
XX
//
See also: output
Jaspar
| Applies to: | Motif and Motif Collection |
Example:
A [1 2 3 0 5 0 0 0 0 0 0 1 ]
C [2 1 0 5 0 0 1 0 0 1 2 0 ]
G [2 2 1 0 0 4 4 0 5 2 0 3 ]
T [0 0 1 0 0 1 0 5 0 2 3 1 ]
>M00002
A [ 4 2 3 2 0 11 0 1 0 0 0 1 1 1 1 ]
C [ 4 5 2 0 11 0 0 2 0 0 0 4 6 4 4 ]
G [ 3 4 4 9 0 0 11 8 0 11 4 3 2 4 2 ]
T [ 0 0 2 0 0 0 0 0 11 0 7 3 2 2 3 ]
Arguments
| Name | Description |
|---|---|
| Format | If the "Default" format is selected, the matrix will be output exactly as it is represented in each motif (which can be either a count matrix, a frequency matrix or a log-odds matrix). However, if the "Frequencies" format is selected, all matrices will be converted to frequency matrices before being output. |
| Header | Specifies which information to include in the header for each motif. The possible options are: include only the motif ID ("ID") or include both the motif ID and motif name (short name) separated with a space ("ID Name"). |
See also: output
XMS
| Applies to: | Motif and Motif Collection |
Example:
<motif>
<name>M00799</name>
<weightmatrix alphabet="DNA" columns="7">
<column pos="0">
<weight symbol="adenine">0.0</weight>
<weight symbol="cytosine">1.0</weight>
<weight symbol="guanine">0.0</weight>
<weight symbol="thymine">0.0</weight>
</column>
<column pos="1">
<weight symbol="adenine">0.9523809523809523</weight>
<weight symbol="cytosine">0.0</weight>
<weight symbol="guanine">0.0</weight>
<weight symbol="thymine">0.047619047619047616</weight>
</column>
<column pos="2">
<weight symbol="adenine">0.0</weight>
<weight symbol="cytosine">1.0</weight>
<weight symbol="guanine">0.0</weight>
<weight symbol="thymine">0.0</weight>
</column>
<column pos="3">
<weight symbol="adenine">0.0</weight>
<weight symbol="cytosine">0.09523809523809523</weight>
<weight symbol="guanine">0.8571428571428571</weight>
<weight symbol="thymine">0.047619047619047616</weight>
</column>
<column pos="4">
<weight symbol="adenine">0.0</weight>
<weight symbol="cytosine">0.0</weight>
<weight symbol="guanine">0.047619047619047616</weight>
<weight symbol="thymine">0.9523809523809523</weight>
</column>
<column pos="5">
<weight symbol="adenine">0.047619047619047616</weight>
<weight symbol="cytosine">0.0</weight>
<weight symbol="guanine">0.9047619047619048</weight>
<weight symbol="thymine">0.047619047619047616</weight>
</column>
<column pos="6">
<weight symbol="adenine">0.0</weight>
<weight symbol="cytosine">0.38095238095238093</weight>
<weight symbol="guanine">0.38095238095238093</weight>
<weight symbol="thymine">0.23809523809523808</weight>
</column>
</weightmatrix>
</motif>
</motifset>
See also: output
MEME_Minimal_Motif
| Applies to: | Motif and Motif Collection |
The format contains the following sections:
- Version (required)
- Alphabet (recommended)
- Strands (optional)
- Background frequencies (recommended)
- Motifs (required)
- Motif name (required)
- Motif letter-probability matrix (recommended*)
- Motif log-odds matrix (optional*)
- Motif URL (optional)
A file in MEME Minimal Motif format must start with the MEME version line which looks like this:
The alphabet line specifies what alphabet to expect the motifs to be in. For DNA motifs this line will be
The strands line indicates if motifs were created from sites on both the given and the reverse complement strands of the DNA sequences.
The background frequencies describe how prevalent each letter of the motif alphabet was in the source sequences which were used to create the motifs. Programs in the MEME Suite use this background to convert between motif letter-probability matrices and log-odds matrices. For DNA alphabets the format is as follows:
A <A-frequency> C <C-frequency> G <G-frequency> T <T-frequency>
A motif name line indicates the start of a new motif and designates an identifier for it which much be unique to the file. It also allows for an (optional) alternate name which does not have to be unique.
The letter probability matrix is a table of probabilities where the rows are positions in the motif and the columns are letters in the alphabet. The columns are ordered alphabetically so for DNA the first column is A, the second is C, the third is G and the last is T. As each row contains the probability of each letter in the alphabet the probabilities in the row must sum to 1. If this section is not specified then the log-odds matrix must be specified.
... (letter-probability matrix goes here) ...
All the "key= value" pairs after the "letter-probability matrix:" text are optional. The "alength= alphabet length" and "w= motif length" can be derived from the matrix if they are not specified, provided there is an empty line following the letter probability matrix. The "nsites= source sites" will default to 20 if it is not provided and the "E= source E-value" will default to zero. The source sites is used to apply pseudocounts to the motif and the source E-value is used for filtering the motifs input to some MEME Suite programs.
Example
ALPHABET= ACGT
strands: + -
Background letter frequencies
A 0.303 C 0.183 G 0.209 T 0.306
MOTIF crp
letter-probability matrix: alength= 4 w= 19 nsites= 17 E= 4.1e-009
0.000000 0.176471 0.000000 0.823529
0.000000 0.058824 0.647059 0.294118
0.000000 0.058824 0.000000 0.941176
0.176471 0.000000 0.764706 0.058824
0.823529 0.058824 0.000000 0.117647
0.294118 0.176471 0.176471 0.352941
0.294118 0.352941 0.235294 0.117647
0.117647 0.235294 0.352941 0.294118
0.529412 0.000000 0.176471 0.294118
0.058824 0.235294 0.588235 0.117647
0.176471 0.235294 0.294118 0.294118
0.000000 0.058824 0.117647 0.823529
0.058824 0.882353 0.000000 0.058824
0.764706 0.000000 0.176471 0.058824
0.058824 0.882353 0.000000 0.058824
0.823529 0.058824 0.058824 0.058824
0.176471 0.411765 0.058824 0.352941
0.411765 0.000000 0.000000 0.588235
0.352941 0.058824 0.000000 0.588235
MOTIF lexA
letter-probability matrix: alength= 4 w= 18 nsites= 14 E= 3.2e-035
0.214286 0.000000 0.000000 0.785714
0.857143 0.000000 0.071429 0.071429
0.000000 1.000000 0.000000 0.000000
0.000000 0.000000 0.000000 1.000000
0.000000 0.000000 1.000000 0.000000
0.000000 0.000000 0.000000 1.000000
0.857143 0.000000 0.071429 0.071429
0.000000 0.071429 0.000000 0.928571
0.857143 0.000000 0.071429 0.071429
0.142857 0.000000 0.000000 0.857143
0.571429 0.071429 0.214286 0.142857
0.285714 0.285714 0.000000 0.428571
1.000000 0.000000 0.000000 0.000000
0.285714 0.214286 0.000000 0.500000
0.428571 0.500000 0.000000 0.071429
0.000000 1.000000 0.000000 0.000000
1.000000 0.000000 0.000000 0.000000
0.000000 0.000000 0.785714 0.214286
See also: output
Motif_Properties
| Applies to: | Motif and Motif Collection |
Example of properties output with the format string "ID,Short name,Classification,Consensus" (with header)
M00006 V$MEF2_01 4.4.1.1 CTCTAAAAATAACyCy
M00005 V$AP4_01 1.3.1.4 wGAryCAGCTGyGGnCnk
M00008 V$SP1_01 2.3.1.0 GGGGCGGGGT
M00007 V$ELK1_01 3.5.2.0 nAAACmGGAAGTnCGT
M00002 V$E47_01 1.2.1.0 vsnGCAGGTGknCnn
M00001 V$MYOD_01 1.2.2.0 srACAGGTGkyG
M00004 V$CMYB_01 3.5.1.1 nCnrnnGrCnGTTGGkGG
M00003 V$VMYB_01 3.5.1.1 AATAACGGnA
Arguments
| Name | Description |
|---|---|
| Format |
This parameter specifies which motif properties to include in the output
as an ordered, comma-separated list of property names.
Note that standard properties are case-insensitive, but user-defined
properties are case-sensitive! Standard motif properties include:
When importing motifs from an existing file, the format parameter should list the properties in the order they appear in the file. If the file contains more properties than you want to import, you are allowed to specify only a subset. For example, if the file has the following five properties ID, Short name, Consensus, Quality and Organisms (in that order), and you only want to import the first three, you can just set the format parameter to "ID, Short name, Consensus" and the remaining properties will simply be ignored. You can also skip properties in the middle of the list by replacing the property name with an asterisk. To import only the properties ID, Consensus and Organism from the mentioned file (i.e. only columns 1,3 and 5), set the format parameter to "ID,*,Consensus,*,Organisms". If the file contains a "header" in the first line (starting with # and describing all the properties), you can use this header rather than naming the properties explicitly by setting the format parameter to a single asterisk (*). Note that the ID property must always be included or MotifLab will complain. |
| Separator | This parameter specifies how to separate the motif properties on each line in the output. The character chosen as the separator will replace the commas in the format string above. Valid options for the separator are "TAB" (default), "Comma", "Semicolon", "Colon" and "Vertical bar". |
| List-separator | Some motif properties may be represented by lists (e.g. binding factors and alternative motif models). The "List-separator" parameter specifies how to separate the entries in such lists. Valid options are "Comma" (default), "TAB", "Semicolon", "Colon" and "Vertical bar". Note that this separator should be different from the separator used to separate the motif properties that are output. |
| Header | If this option is selected, a header line starting with # will be included at the beginning of the output. The header line describes which motif properties are included. |
See also: output, HTML_MotifTable, Module_Properties, Sequence_Properties, Properties
Module_Properties
| Applies to: | Module and Module Collection |
Example of properties output with the format string "ID, Size, Max IC, Motifs" (with header)
MOD0040 3 43.5204389330021 NFKAPPAB65,SP1,EGR2
MOD0078 3 54.12787872722569 STAT,TITF1,STAT3
MOD0081 3 39.159961609322714 P300,STAT,IRF7
MOD0069 6 95.6314848710252 AP1FJ,STAT,AP1,CREBP1CJUN,STAT1,STAT3
MOD0070 4 76.28839485773265 STAT,IRF1,STAT1,ISRE
MOD0101 2 42.324578249877625 NRSE,MTATA
MOD0105 2 38.59871626791364 PAX5,MTATA
Arguments
| Name | Description |
|---|---|
| Format |
This parameter specifies which module properties to include in the output
as an ordered, comma-separated list of property names.
Note that standard properties are case-insensitive, but user-defined
properties are case-sensitive! Standard module properties include:
|
| Separator | This parameter specifies how to separate the module properties on each line in the output. The character chosen as the separator will replace the commas in the format string above. Valid options for the separator are "TAB" (default), "Comma", "Semicolon", "Colon" and "Vertical bar". |
| List-separator | Some smodule properties may be represented by lists (e.g. GO-terms). The "List-separator" parameter specifies how to separate the entries in such lists. Valid options are "Comma" (default), "TAB", "Semicolon", "Colon" and "Vertical bar". Note that this separator should be different from the separator used to separate the module properties that are output. |
| Header | If this option is selected, a header line starting with # will be included at the beginning of the output. The header line describes which module properties are included. |
See also: output, Motif_Properties, Sequence_Properties, Properties, HTML_ModuleTable
HTML_MotifTable
| Applies to: | Motif and Motif Collection |
Arguments
| Name | Description |
|---|---|
| Format |
This parameter specifies which motif properties to include in the output
as an ordered, comma-separated list of property names.
Note that standard properties are case-insensitive, but user-defined
properties are case-sensitive! Standard motif properties include:
|
| Sequence logo height | The height of the sequence logos (if logos are included). |
| Sequence logo width | The maximum width of sequence logos. If this is specified, logos will be scaled to fit within this limit when necessary. A value of 0 means no limit. |
| Sequence logos | Specifies whether the image files for motif sequence logos should be made specifically for this output data object and given file names reflecting this fact ("New images") or if the image files should be named after the motifs themselves (e.g. "M00134.gif") and allowed to be shared by other HTML-formatted output data objects that also contain sequence logos for the same motifs ("Shared images"). |
| multiline | Some motif properties may be represented by lists (e.g. binding factors and alternative motif models). If the "multiline" option is selected, these lists will be broken across multiple lines in a table cell. If the "multiline" option is not selected, these properties will be output as comma-separated lists. |
| Headline | An optional headline which will be displayed above the table. |
See also: output, Motif_Properties
HTML_ModuleTable
| Applies to: | Module and Module Collection |
Arguments
| Name | Description |
|---|---|
| Format |
This parameter specifies which module properties to include in the output
as an ordered, comma-separated list of property names.
Note that standard properties are case-insensitive, but user-defined
properties are case-sensitive! Standard module properties include:
|
| Logo max width | The maximum width of module logos. If this is specified, logos will be scaled to fit within this limit when necessary. A value of 0 means no limit. |
| Logos | Specifies whether the image files for module logos should be made specifically for this output data object and given file names reflecting this fact ("New images") or if the image files should be named after the modules themselves (e.g. "MOD0003.gif") and allowed to be shared by other HTML-formatted output data objects that also contain logos for the same modules ("Shared images"). A third option is to output logos using textual rather than graphical representations ("Text"). |
| multiline | Some module properties may be represented by lists. If the "multiline" option is selected, these lists will be broken across multiple lines in a table cell. If the "multiline" option is not selected, these properties will be output as comma-separated lists. |
| Headline | An optional headline which will be displayed above the table. |
See also: output
BindingSequences
| Applies to: | Motif and Motif Collection |
When the format is used for output, it will output a header-line for each motif followed by a list of all the binding sequences associated with that motif. If the motif has no annotated binding sequences, it can either output an IUPAC consensus sequence or, optionally, a set of randomly generated binding sequences that taken together will approximate the base frequencies of the motif binding matrix to a given precision.
Example:
CACGTG
CAgGTG
CACGTG
CACGTG
CcCGTG
CACGaG
CACGTG
>Motif2
nrATGAyvTA
>Motif3 Unknown
AGCTACT
AGCTAGT
GGCTAGT
AGCTAGT
aGCTACT
AGCTAGT
AGCTAGG
#Motif with separate headers for each binding sequence
>Motif4-1
AGCTACT
>Motif4-2
AGCTAGT
>Motif4-3
GGCTAGT
>Motif4-4
Arguments
| Name | Description |
|---|---|
| Binding sequence property | This parameter specifies the name of the user-defined motif property that contains (or will contain) the binding sequences as a comma-separated list. If no property is specified when importing motifs from file, the binding sequences will not be stored together with the motif. If no property is specified when outputting motifs, the consensus sequence (or a set of randomly generated sequences) will be output rather than the actual binding sequences. |
| Header | Specifies which information to include in the header for each motif. The possible options are: include only the motif ID ("ID") or include both the motif ID and motif name (short name) separated with a space ("ID Name"). |
| Separate headers | If this option is selected, each binding sequence will be preceded by a separate header on the previous line. These headers will be similar to the regular headers except that the motif ID will be immediately followed by a hyphen and an incremental number. |
| When missing |
This parameter controls what to output when a motif does not have any
annotated binding sequences associated with it.
|
| Random sequences precision | This advanced parameter controls the number of sequences that will be randomly generated if the "When missing" parameter is set to "Generate random" and the binding matrix of a motif is a frequency matrix (or log-transformed matrix) rather than a count matrix. The value N (between 1 and 6) of this parameter is directly related to the number of sequences output (=10^(N-1)) and hence also the number of decimals (N-1) used for the frequencies if a frequency matrix is recreated from these binding sequences. |
See also: output
Background formats
PriorityBackground
| Applies to: | Background Model |
The background model's order (k) may be any integer between 0 and 5. For a k-th order model the file must contain exactly
For example for a 3rd order model the numbers represent:
P(A)
P(C)
P(G)
P(T)
P(A|A)
P(C|A)
P(G|A)
P(T|A)
P(A|C)
....
P(T|T)
P(A|AA)
P(C|AA)
.....
P(T|TT)
......
P(T|TT)
P(A|AAA)
P(A|AAC)
.......
P(A|TTT)
........
P(T|TTT)
In this case the file must contain 4+16+64+256=340 numbers.
IMPORTANT!!! Notice that every group of 4 consecutive numbers must add up to 1 to form a probability distribution.
See also: output
MEME_Background
| Applies to: | Background Model |
The format for n-order Markov background models is as follows.
The file must contain one line for each combination of 1, 2, ..., n-1 letters in the alphabet. The DNA alphabet is ACGT.
Each line must contain the letter combination followed by the letter combination's frequency (probability). All other lines in the file are ignored, including comment lines starting with '#'.
For example, a 0-order Markov model file might contain:
a 0.324
c 0.176
g 0.176
t 0.324
A 1st-order Markov model file might contain:
a 0.324
c 0.176
g 0.176
t 0.324
# tuple frequency_non_coding
aa 0.119
ac 0.052
ag 0.056
at 0.097
ca 0.058
cc 0.033
cg 0.028
ct 0.056
ga 0.056
gc 0.035
gg 0.033
gt 0.052
ta 0.091
tc 0.056
tg 0.058
tt 0.119
See also: output
INCLUSive_Background_Model
| Applies to: | Background Model |
The file starts with a comment line which identifies the format (#INCLUSive Background Model v1.0).
Next follows a description of the order of the background model (#Order) and two informational fields describing respectively a genome identifier (#Organism) and the path referring to the sequences data where the model is extracted from (#Sequences).
The single nucleotide frequencies for A,C,G,T are described by 4 tab separated values (between 0 and 1) on the line following #snf. They represent the probability (Pr) to find the respective nucleotide in the sequence dataset where the background is modelled for, independent of the position of this nucleotide in the sequences.
Pr(A) Pr(C) Pr(G) Pr(T)
The section following #oligo describes the probability of all possible combinations of the nucleotides A,C,G,T of length equal to the background model order (also called an oligonucleotide) in the sequence dataset where the background is modelled for. The total number of oligonucleotides are printed on separate lines and equals 4 powered to the background model order (e.g. 16 for a second order model). The section starts with the oligonucleotide consisting of all A, followed by oligonucleotides where each next position in the oligonucleotide A is repeatedly replaced by respectively C,G,T. Below example is for a second order background model.
Pr(AA)
Pr(AC)
Pr(AG)
Pr(AT)
Pr(CA)
Pr(CC)
Pr(CG)
Pr(CT)
Pr(GA)
Pr(GC)
Pr(GG)
Pr(GT)
Pr(TA)
Pr(TC)
Pr(TG)
Pr(TT)
The higher-order background model is described in the section following #transition matrix. Each line in this section describes the tab separated probabilities (Pr) of finding nucleotide A respectively C, G and T given a set of preceding nucleotides of length equal to the background model order. The total number of lines equals 4 powered to the background model order. The preceding oligonucleotide for the first line consists of all A, and in next lines A is repeatedly replaced by respectively C,G,T on each next position in the oligonucleotide. Below example is for a second order background model.
Pr(A|AA) Pr(C|AA) Pr(G|AA) Pr(T|AA)
Pr(A|AC) Pr(C|AC) Pr(G|AC) Pr(T|AC)
Pr(A|AG) Pr(C|AG) Pr(G|AG) Pr(T|AG)
Pr(A|AT) Pr(C|AT) Pr(G|AT) Pr(T|AT)
Pr(A|CA) Pr(C|CA) Pr(G|CA) Pr(T|CA)
Pr(A|CC) Pr(C|CC) Pr(G|CC) Pr(T|CC)
Pr(A|CG) Pr(C|CG) Pr(G|CG) Pr(T|CG)
Pr(A|CT) Pr(C|CT) Pr(G|CT) Pr(T|CT)
Pr(A|GA) Pr(C|GA) Pr(G|GA) Pr(T|GA)
Pr(A|GC) Pr(C|GC) Pr(G|GC) Pr(T|GC)
Pr(A|GG) Pr(C|GG) Pr(G|GG) Pr(T|GG)
Pr(A|GT) Pr(C|GT) Pr(G|GT) Pr(T|GT)
Pr(A|TA) Pr(C|TA) Pr(G|TA) Pr(T|TA)
Pr(A|TC) Pr(C|TC) Pr(G|TC) Pr(T|TC)
Pr(A|TG) Pr(C|TG) Pr(G|TG) Pr(T|TG)
Pr(A|TT) Pr(C|TT) Pr(G|TT) Pr(T|TT)
Example
#
#Order = 1
#Organism = Human
#Sequences =
#
#snf
0.2570.25340.24650.2432
#oligo frequency
0.257
0.2534
0.2465
0.2432
#transition matrix
0.3121 0.1944 0.2751 0.2184
0.2751 0.3014 0.1547 0.2688
0.24 0.2718 0.2943 0.1939
0.197 0.2469 0.2637 0.2924
See also: output
Other formats
MapFormat
| Applies to: | Map |
Arguments
| Name | Description |
|---|---|
| Entry separator | This parameter specifies which character to use to separate the key-value entries from each other in the output. The default is to output one entry on each line, but other means of separation is also possible. Valid values for this parameter are "Newline", "Space", "Tab", "Comma", "Semicolon" and "Colon". Note that the separator chosen here must be different from the "Key-value separator" selected below. |
| Key-value separator | This parameter specifies which character to use to separate the key from the value for each entry. The default separation character is the equals sign (=), but other options are also available. Valid values for this parameter are "Newline", "Space", "Tab", "Comma", "Semicolon" and "Colon". Note that the separator chosen here must be different from the "Entry separator" selected above. |
| Include entries | A collection which specifies which entries to include in the output. If no collection is specified (or an incompatible collection is chosen) all the entries will be output. |
| Include default | If this option is selected, an entry for the default value will be included in the output. This will usually have the key "_DEFAULT_" (or no key at all). |
See also: MapExpression, output, Numeric Maps
MapExpression
| Applies to: | Map |
When outputting a Map in MapExpression format, the expression parameter should be a string which contains two special field codes: {KEY} and {VALUE} (the braces must be included and the letters must be in uppercase). The KEY field will be replaced by the name (identifier) of the data object in the map (motif, module or sequence) and the VALUE field will be replaced by the corresponding value for this data object. For example, the expression "{KEY}={VALUE}" will output the name of the data object and the value separated by an equals-sign, whereas the expression "ENTRY\t{VALUE}\t{KEY}" will output three TAB-separated columns on each row where the first column always has the text "ENTRY", the second column is the value and the last column is the name of the data object.
When importing a file in MapExpression format, the expression should be a regular expression string (formatted according to JAVA regex rules) containing at least two "capturing groups" enclosed in parenthesis. The two capturing groups should match the data name (key) and value respectively. The integer parameters "Key group" and "Value group" are used to tell MotifLab which of the groups are associated with each of these fields. For example, if the entries in the file correspond to the (output) expression "{KEY}={VALUE}", then the input expression could be "(\S+?)=(\S+)" with the value of "Key group" set to 1 and "Value group" set to 2. If the file is in the format "ENTRY\t{VALUE}\t{KEY}", then the input expression "ENTRY\t(\S+)\t(\S+)" can be used with "Key group" set to 2 and "Value group" set to 1 (since the key now occurs after the value in each line). It is possible to use more than two capturing groups, and the "Key group" and "Value group" parameters must then be adjusted accordingly.
Note that double quotes should preferably be avoided in the expression string since this can lead to parsing problems in the current version of MotifLab.
Arguments
| Name | Description |
|---|---|
| Expression | A string which defines the format to use for output or input. When outputting data in MapExpression format, this expression string should contain the two special field codes: {KEY} and {VALUE}, and these will be replaced by key-value data from the map. When reading input in MapExpression format, the expression string should be a regular expression containing at least two "capturing groups" that can capture the key and value information respectively from each line in the input. The parameters "Key group" and "Value group" below specify which capturing groups should be used to capture these two properties. |
| Key group | An integer number that specifies which capturing group that captures the "key" from the input. This parameter is only applicable when reading input in the MapExpression format. |
| Value group | An integer number that specifies which capturing group that captures the "value" from the input. This parameter is only applicable when reading input in the MapExpression format. |
| Include entries | A collection which specifies which entries to include in the output. If no collection is specified (or an incompatible collection is chosen) all the entries will be output. |
| Include default | If this option is selected, an entry for the default value will be included in the output. This will usually have the key "_DEFAULT_" (or no key at all). |
See also: MapFormat, output, Numeric Maps
ExcelMap
| Applies to: | Map |
Arguments
| Name | Description |
|---|---|
| Key column | This parameter specifies the column that contains the keys (names of motifs, modules or sequences). The column is specified as an integer number starting at 1 for the first (leftmost) column in the Excel file (first sheet). |
| Value column | This parameter specifies the column that contains the corresponding map values for the motifs/modules/sequences. The column is specified as an integer number starting at 1 for the first (leftmost) column in the Excel file (first sheet). |
| Include entries | A collection which specifies which entries to include in the output. If no collection is specified (or an incompatible collection is chosen) all the entries will be output. |
| Include default | If this option is selected, an entry for the default value will be included in the output. This will usually have the key "_DEFAULT_" (or no key at all). |
See also: MapFormat, MapExpression, ExcelProfile, output, Numeric Maps
ExpressionProfile
| Applies to: | Expression Profile |
Arguments
| Name | Description |
|---|---|
| Sequence name delimiter | This parameter specifies which character to use to separate the sequence names (first column) from the expression data values (the rest of the columns). Valid values for this parameter are "Space", "Tab", "Comma", "Semicolon", "Equals" and "Colon". |
| Condition delimiter | This parameter specifies which character to use to separate the expression data columns from each other (the different conditions). Valid values for this parameter are "Space", "Tab", "Comma", "Semicolon" and "Colon". |
| Header | Specifies whether a descriptive header should be included as the first line in the output. The parameter value "None" will not output any header. The value "Column names" will output a headder containing the name of each column (separated by the same characters as the data on the following lines). The value "#Column names" will output the same header as the previous setting, except that the line will be preceeding by a # sign (which will usually be interpreted as a comment line). |
See also: ExcelProfile, output, Expression Profile
ExcelProfile
| Applies to: | Expression Profile |
Arguments
| Name | Description |
|---|---|
| Key column | This input parameter specifies the column that contains the sequence names. The column is specified as an integer number starting at 1 for the first (leftmost) column in the Excel file (first sheet). |
| Value columns | This optional input parameter specifies which columns that contain the expression data that should be included in the expression profile. This can be specified as a comma-separated list of column indices (starting at 1 for the leftmost column in the Excel file). It is also possible to specify ranges of columns using a hyphen. For example, if this parameter is set to "2,3,5-7,9" the profile will import data from the columns 2,3,5,6,7 and 9 (note that column 6 is in the range 5-7). If this parameter is left blank, all columns with valid numeric values to the right of the key column (containing the sequence names) will be included. |
| Include entries | A sequence collection which specifies which sequences to include in the output. If no collection is specified, all sequences will be output. |
| Header | If this parameter is selected when outputting data, a header containing the names of the condition columns will be output as the first row in the Excel file. If selected when importing data, the values found in the first row of the Excel file will be used as condition headers. |
See also: output, Expression Profile
HTML
| Applies to: | Analysis |
Arguments
| Name | Description |
|---|---|
| analysis-specific | Each type of analysis can have its own output parameters for the HTML format, but common parameters include which entries to include in the output (often specified as a Collection) and how to sort the output. |
| Logos |
Analyses related to motifs or modules usually allow you to include a
graphical representation of each motif or module in the output. This is
called a "logo", and you have a few options regarding how this will be
handled.
|
See also: output, Analysis
Excel
| Applies to: | Analysis |
Arguments
| Name | Description |
|---|---|
| analysis-specific | Each type of analysis can have its own output parameters for the Excel format, but common parameters include which entries to include in the output (often specified as a Collection) and how to sort the output. |
See also: output, Analysis
RawData
| Applies to: | Analysis |
Arguments
| Name | Description |
|---|---|
| analysis-specific | Each type of analysis can have its own output parameters for the RawData format, but common parameters include which entries to include in the output (often specified as a Collection) and how to sort the output. |
See also: output, Analysis
Template
| Applies to: | Text Variable |
The Template format (and its companion TemplateHTML) can be used in such cases to achieve greater control over the presentation of the output. The data formats requires a "template" document which must be provided in the form of a Text Variable. This template can contain regular text which can be interspersed with references to named data objects on the form: {dataobject}. When the template Text Variable is output in Template-format, the references to data objects in the text will be replaced with the actual contents of these data objects. No error message or warning will be given if the data object named in the reference does not exist or if there is some other mistake in the reference. In such cases, MotifLab will simply leave the original reference as is in the output. Note that only "simple" data objects such as Numeric Variables, Text Variables, Collections and OutputData objects can be referenced directly in the template. However, other types of data (such as feature data, partitions or maps) can be included by first outputting the original data object to an intermediate OutputData object in a selected data format. This intermediate object can then be referenced in the template.
The contents of a referenced data object will usually be included directly in a default format, but some control over the presentation is provided with additional options that can be specified after the name of the data object in the reference following a colon: {dataobject:options}.
Some data types can even have multiple options like so: {dataobject:option1:option2:etc...}
Available format-options for different data types:
| Data type | Option | Description |
|---|---|---|
| Numeric Variable | Precision | The number of decimals
to include for real values in the output can be specified in the
option. For example, to output the variable X with 2 decimals use the reference code: {X:2}. |
| Percentage | If the Numeric
Variable holds a value between 0 and 1.0 which should be output as a
percentage number, simply place a percentage sign in the option. The
value of the variable will then automatically be multiplied by 100 and
suffixed with a %-sign in the output. This option can be combined with
the precision option by writing the %-sign after the number of
decimals. If you want a space between the value and the %-sign in the
output you can place the space-code "\s" before the
%-sign (or if you combine precision number with the %-option you can just
place a normal space between them). Examples: The following shows different output for the variable X=0.2394278. The reference code {X:%} will output 23.94278% The reference code {X:\s%} will output 23.94278 % The reference code {X:0%} will output 23% The reference code {X:2%} will output 23.94% The reference code {X:3 %} will output 23.942 % |
|
| Collection | Separator | Collections are normally output as sorted lists with entries separated by commas. However, it is also possible to specify a different separator as an option. E.g. to output the collection JASPAR with entries separated by semicolons instead of commas, use the reference code {JASPAR:;}. A space-separator can be specified as "\s", TAB-separator as "\t" and linebreaks as "\n" for plain text or "<br>" for HTML. To use a colon as a separator, simply type "colon", e.g. {JASPAR:colon}. |
| Size | To output the size of a collection rather than listing the entries, use the "size" option, or equivalently enclose the name of the collection in vertical bars. E.g. {JASPAR:size} or {|JASPAR|}. | |
| Text Variable and Output Data |
Separator | The contents of Text Variables and Output Data objects are usually included verbatim in the final document one line at a time. However, it is possible to use a different separator for the lines rather than the usual linebreak separator (which is "\n" for plain text and "<br>" for HTML). This separator is then specified as the first option. Spaces, TABs and colons can be used as separators as described for the Collections type above. |
| Sorting | It is possible to
sort the lines from the Text Variable or Output Data according to a
natural order by specifying a second option (after the first "separator"
option). There are many ways to do this. If the second option
includes the text "sort", "asc" or "A" the lines will be sorted in ascending
order (e.g. "sort", "sorted", "sort ascending", "ascend","sortA" or
simply "A"). However, if the option contains the text "des" or "D" they
will be sorted in descending order (e.g. "sort desc", "sort
descending", "descend", "desc", "sortD" or simply "D").
The following examples show how the lines from the Text Variable X can be sorted. Note that since sorting must be specified as a second option, the "separator" must be included as the first option (but by just leaving it blank the default separator will be used). The reference code {X::sort} will output X with lines in ascending order separated by default linebreaks. The reference code {X::ascending} will output X with lines in ascending order separated by default linebreaks. The reference code {X::D} will output X with lines in descending order separated by default linebreaks. The reference code {X:\t:asc} will output X with lines in ascending order separated by TABS. |
|
| Remove duplicates |
The second option can also be used to specify that duplicate lines in
the Text Variable or Output Data object should be removed by
including the text "uniq" or "U" anywhere in the option. Note that
this can be combined with the sorting option, as shown in the last
three examples below: The reference code {X::uniq} will output all unique lines from X separated by default linebreaks. The reference code {X::sorted unique} will output all unique lines from X sorted in ascending order. The reference code {X::unique ascending} will output all unique lines from X sorted in ascending order. The reference code {X:,:DU} will output all unique lines from X in descending order separated with commas. |
Example:
In the following scenario, a user has created a protocol script to find significant transcription factor motifs in promoters for sets of sequences that are either up- or downregulated at three different timepoints (1h, 2h or 3h). The sequence collections are named Up1, Up2, Up3, Down1, Down2 and Down3, and the collections of significant motifs are called Motifs_Up1, Motifs_Up2, Motifs_Up3, Motifs_Down1, Motifs_Down2 and Motifs_Down3. What the user wants now is to create a simple report that contains information about how many sequences there were in each such collection and list the names of the significant TFs for each collection. The following protocol first creates six OutputData objects containing names of significant TFs and then creates the final report based on a predefined template.
Motifs_Up1_out = output Motifs_Up1 in Motif_Properties format {Format="Clean Short name"}
Motifs_Up2_out = output Motifs_Up2 in Motif_Properties format {Format="Clean Short name"}
Motifs_Up3_out = output Motifs_Up3 in Motif_Properties format {Format="Clean Short name"}
Motifs_Down1_out = output Motifs_Down1 in Motif_Properties format {Format="Clean Short name"}
Motifs_Down2_out = output Motifs_Down2 in Motif_Properties format {Format="Clean Short name"}
Motifs_Down3_out = output Motifs_Down3 in Motif_Properties format {Format="Clean Short name"}
TemplateText = new Text Variable(File:"Template_text.txt", format=Plain)
Output1= output TemplateText in Template format
The file "Template_text.txt" which is used in the protocol above to provide the template text for the final report is shown below. All references to data objects in the template are marked in red color. First the template produces a 2x3-table with the sizes of each of the sequence collections. Then it writes out 6 lines listing the significant transcription factors for each such collection. It would be possible to reference the motif collections directly in the template, e.g. {Motifs_Up1}. However, this would then only list the IDs of the motifs and not the TF-names, so instead the protocol above uses the Motif_Properties data format to output the "clean short name" of each motif in each collection to an intermediate OutputData object that is referenced instead (Motifs_Up1_out). These OutputData objects contains one TF-name on each line and there might be duplicate names if there are several motif models for the same TF. Hence, the template specifies that the names in the OutputData objects should be sorted alphabetically and duplicate names should be removed (using the "sorted unique" option). Also, rather than using a linebreak to separate the TF-names, a comma should be used instead.
Number of genes up- and down-regulated at different time points:
Time 1h 2h 3h
--------------------------------
UP {Up1:size} {Up2:size} {Up3:size}
DOWN {Down1:size} {Down2:size} {Down3:size}
================================
Significant transcription factors in promoters of these genes:
Up 1h: {Motifs_Up1_out:,:sorted unique}
Down 1h: {Motifs_Down1_out:,:sorted unique}
Up 2h: {Motifs_Up2_out:,:sorted unique}
Down 2h: {Motifs_Down2_out:,:sorted unique}
Up 3h: {Motifs_Up3_out:,:sorted unique}
Down 3h: {Motifs_Down3_out:,:sorted unique}
The result could look something like this:
Number of genes up- and down-regulated at different time points: Time 1h 2h 3h -------------------------------- UP 23 42 31 DOWN 17 35 26 ================================ Significant transcription factors in promoters of these genes: Up 1h: EGR1,FOS1 Down 1h: Up 2h: FOXD1,GATA2,HOXA5,IRX5,JUND Down 2h: E2F5,KLF15,NR2C1 Up 3h: ATF5,NFX1,NKX3-1,ZHX2 Down 3h: EVI1,HEYL,HOXC9,POU2F1
See also: TemplateHTML, output, Text Variable, Output Data
TemplateHTML
| Applies to: | Text Variable |
The TemplateHTML format (and its companion Template) can be used in such cases to achieve greater control over the presentation of the output. The data format requires a "template" document which must be provided in the form of a Text Variable. This template can contain regular text and HTML-markup which can be interspersed with references to named data objects on the form: {dataobject}. When the template Text Variable is output in TemplateHTML-format, the references to data objects in the text will be replaced with the actual contents of these data objects. No error message or warning will be given if the data object named in the reference does not exist or if there is some other mistake in the reference. In such cases, MotifLab will simply leave the original reference as is in the output. Note that only "simple" data objects such as Numeric Variables, Text Variables, Collections and OutputData objects can be referenced in the template. However, other types of data objects (such as feature data, partitions or maps) can be included by first outputting these to intermediate OutputData objects which can then be referenced.
The contents of a referenced data object will usually be included directly in a default format, but some control over the presentation is provided with additional options which are described in the documentation for the Template format.
Example:
In the following scenario, a user has created a protocol script to find significant transcription factor motifs in promoters for sets of sequences that are either up- or downregulated at three different timepoints (1h, 2h or 3h). The sequence collections are named Up1, Up2, Up3, Down1, Down2 and Down3, and the collections of significant motifs are called Motifs_Up1, Motifs_Up2, Motifs_Up3, Motifs_Down1, Motifs_Down2 and Motifs_Down3. What the user wants now is to create a simple report that contains information about how many sequences there were in each such collection and list the names of the significant TFs for each collection. The following protocol first creates six OutputData objects containing names of significant TFs and then creates the final report based on a predefined template.
Motifs_Up1_out = output Motifs_Up1 in Motif_Properties format {Format="Clean Short name"}
Motifs_Up2_out = output Motifs_Up2 in Motif_Properties format {Format="Clean Short name"}
Motifs_Up3_out = output Motifs_Up3 in Motif_Properties format {Format="Clean Short name"}
Motifs_Down1_out = output Motifs_Down1 in Motif_Properties format {Format="Clean Short name"}
Motifs_Down2_out = output Motifs_Down2 in Motif_Properties format {Format="Clean Short name"}
Motifs_Down3_out = output Motifs_Down3 in Motif_Properties format {Format="Clean Short name"}
TemplateText = new Text Variable(File:"Template_text.html", format=Plain)
Output1= output TemplateText in TemplateHTML format
The file "Template_text.html" which is used in the protocol above to provide the template text for the final report is shown below. All references to data objects in the template are marked in red color. First the template produces a 2x3-table with the sizes of each of the sequence collections. Then it writes out a second table listing the significant transcription factors for each such collection. It would be possible to reference the motif collections directly in the template, e.g. {Motifs_Up1}. However, this would then only list the IDs of the motifs and not the TF-names, so instead the protocol above uses the Motif_Properties data format to output the "clean short name" of each motif in each collection to an intermediate OutputData object that is referenced instead (Motifs_Up1_out). These OutputData objects contains one TF-name on each line and there might be duplicate names if there are several motif models for the same TF. Hence, the template specifies that the names in the OutputData objects should be sorted alphabetically and duplicate names should be removed (using the "AU" option).
<h2>Number of genes up- and down-regulated at different time points:</h2>
<table>
<tr><th>Time</th><th>1h</th><th>2h</th><th>3h</th></tr>
<tr><td>Up</td><td>{Up1:size}</td><td>{Up2:size}</td><td>{Up3:size}</td></tr>
<tr><td>Down</td><td>{Down1:size}</td><td>{Down2:size}</td><td>{Down3:size}</td></tr>
</table>
<br>
<h2>Significant transcription factors in promoters of these genes:</h2>
<table>
<tr>
<th style="background-color:#E0E0E0;">1h</th>
<th style="background-color:#C8C8C8;">2h</th>
<th style="background-color:#B0B0B0;">3h</th>
</tr>
<tr>
<td valign=top style="background-color:#FFD0D0;">{Motifs_Up1_out::AU}</td>
<td valign=top style="background-color:#FFC0C0;">{Motifs_Up2_out::AU}</td>
<td valign=top style="background-color:#FFC0B0;">{Motifs_Up3_out::AU}</td>
</tr>
<tr>
<td valign=top style="background-color:#D0FFD0;">{Motifs_Down1_out::AU}</td>
<td valign=top style="background-color:#C0FFC0;">{Motifs_Down2_out::AU}</td>
<td valign=top style="background-color:#B0FFB0;">{Motifs_Down3_out::AU}</td>
</tr>
</table>
The result could look something like this:
Number of genes up- and down-regulated at different time points:
| Time | 1h | 2h | 3h |
|---|---|---|---|
| Up | 23 | 42 | 31 |
| Down | 17 | 35 | 26 |
Significant transcription factors in promoters of these genes:
| 1h | 2h | 3h |
|---|---|---|
| EGR1 FOS1 |
FOXD1 GATA2 HOXA5 IRX5 JUND |
ATF5 NFX1 NKX3-1 ZHX2 |
| E2F5 KLF15 NR2C1 |
EVI1 HEYL HOXC9 POU2F1 |
See also: Template, output, Text Variable, Output Data
Properties
| Applies to: | Motif, Motif Collection, Module, Module Collection, Sequence and Sequence Collection |
The following example shows a small template to output three motif properties:
Name = {Short name}
Length = {size}
Repeat blocks
A repeat block is a convenient, short-hand way of outputting multiple properties in a similar fashion. A repeat block starts off with a header line consisting of a percentage sign followed by a comma-separated list of properties. This header is then followed by any number of regular template lines. The repeat block is ended when encountering a line containing a double percentage sign. When a data object is output, all the lines between the single and double percentage signs will be repeated once for each property in the list. In additional to regular named property references, the lines within a repeat block can contain the special references "{key}" and "{value}" which refers to the name and value of the property currently being processed. The list of properties to repeat over can contain the wildcard symbol "*", which will then add all known properties to the list, or the subset wildcards "*standard" (all standard properties) and "*user" (all non-standard user-defined properties). If the name of a property in the list is preceeded by a minus sign, it will be removed from the current list of properties. Hence, the list "*,-ID" will include all properties except "ID".
The motif template below will output the properties "Short name", "Consensus" and "IC-content" followed by all user-defined properties except the two named "Pazar" and "Medline". Each line will consist of the ID of the current motif followed by a colon, and then the name of a property from the list and its respective value separated by an equals sign.
{ID}: {key} = {value}
%%
The list of properties in the repeat block header can optionally be followed by a colon and then a series of settings specifications in the form of keywords that further control the formatting of the repeat block:
- skip and blank: These settings will override the "If missing" parameter of the data format for this repeat block only.
A header on the form "%property:skip" thus essentially represents a conditional block that will only be output if the named property is defined for the data object. - [default]: A value enclosed in brackets will be used as a default when a data object does not have a defined value for the property.
If specified, this setting overrides both "skip" and "blank". - "separator": A string enclosed in double quotes will be used as the separator when outputting properties with multiple values (lists).
Use "\t" to represent TAB and "\n" for newline. This setting will default to comma unless specified. - inline: This setting is really only useful if the repeat block consists of a single line. When in use, all the properties will be output on the same line immediately after each other. Also, unless explicitly specified with the "break before/after" settings, there will be no linebreaks occurring before and after the list of properties.
- break before and break after: These keywords can be used in combination with the "inline" setting to force linebreaks to occur before and/or after the inline repeat block.
- 'separator': A string enclosed in single quotes will be used as the separator when outputting a list of properties with an inline repeat block.
Use "\t" to represent TAB. If unspecified, no separator will be output between the properties. - expand list: This keyword (which is incompatible with "inline") can apply when a property has multiple values in the form of an explicit list or a comma-separated string. In this case, instead of replacing the {value} reference with the whole list as one string (with entries separated by commas unless otherwise specified), the whole repeat block will itself be repeated for every value from the list, and each time the {value} reference will be replaced with this single value.
Matrix block
A matrix block is special kind of repeat block used to format the variable-length binding matrix of a motif. The special "{key} and {value}" references are not defined for this kind of repeat block. Instead the matrix block has a a few other references, including "{A}, {C}, {G} and {T}" which refers to the matrix values for these bases. A binding matrix can be output in either vertical (N×4) or horizontal (4×N) orientation, and the repeat block will behave differently in these two cases.
In the case of vertical orientation, the repeat block will be repeated N times (where N is the length of the motif), and with each repetition the values of {A}, {C}, {G} and {T} will refer to the next position in the matrix. The current position (row counter) can be accessed with the special references "{row}" or "{rowXX}", where the latter will always return the position in double digits. (If you want a row counter starting at 0 rather than 1, use the references "{row-1}" or "{rowXX-1}" instead). Another special reference "{x}" can be used to access the consensus base letter for the position. This base letter will be in uppercase for regular DNA bases and in lowercase for degenerate bases. The reference "{X}" will always return the base letter in uppercase.
{row} {A},{C},{G},{T},{x}
%%
In the case of horizontal orientation, the repeat block itself will not be repeated more than once. Instead, the references {A}, {C}, {G}, {T} and {x} will contain lists of values rather than single values. The values in these lists will be separated by a comma unless a different separator is specified within a pair of double quotes in the block header. The special reference "{columns}" (and also "{columnsXX}, {columns-1} and {columnsXX-1}") contains column position counters in similarly formatted lists.
{columns}
{A}
{C}
{G}
{T}
%%
The settings string in the header of the matrix block can contain the keywords "count" or "frequency" to force the matrix to be output in a count-matrix format (only integers) or normalized frequencies format (values between 0 and 1) instead of the default formats used by each motif internally.
Headers, footers and separators
The template can optionally contain the definition of a header that will be output at the beginning of the document, a footer that will be output at the end, and a separator that will be output between each entry when multiple data objects are output. If included, these must be defined at the very top of the template and introduced by the keywords "HEADER:", "FOOTER:" and "SEPARATOR:" respectively, as shown in the example below (note that the keywords have to be in uppercase!). Each definition must be contained on a single line, but you can force linebreaks by including the newline character "\n". The header, footer and separator can not contain any property references, but they can contain the special reference "{size}" which is the size of the collection being output and "{counter}" which is a number that starts at 1 and is incremented for each entry output ("{counter-1}" is similar but starts the counting at 0).
FOOTER:this is the footer text...
SEPARATOR:this line of text will separate the entries
Since templates can contain any text, the "Properties" format can be used to mimic nearly all other data formats for outputting motifs, modules or sequences, as demonstrated by the following example templates:
Example #1: BED format
Since genomic sequence coordinates in BED files start at 0 rather than 1, a "[0]" suffix has been added to the "genomic start" property in the second column. The same suffix has not been added to the "genomic end" property in third column, however, since this coordinate is exclusive according to the BED-format definition, and exclusive 0-indexed end-coordinates are the same as inclusive 1-indexed end-coordinates. The "score" value in column five is just set to the constant number "1000".
Example #2: GFF format
The ninth and final column of the GFF sequence format contains a semi-colon separated list of additional attributes defined as key="value" pairs. The template below outputs the sequence name as the first such attribute and if the sequence has any other user-defined attributes these will be output directly behind on the same line using a repeat block with the "inline" setting.
%*user: inline, skip, break after
{key}="{value}";
%%
Example #3: JASPAR format
The motif format used by the JASPAR database is very simple, consisting of just a FASTA-like header followed by four lines that define the matrix values for the four DNA bases. This template demonstrates how to use a repeat block to output the "matrix" property of the motif in a horizontal orientation. The TAB-sign "\t" is used to separate the columns in the matrix rather than the default separator (comma). Note that the template also includes an explicit entry SEPARATOR which is inserted between every motif in the output. In this case the separator is empty and will result in an empty line being output between each motif.
>{ID}
%matrix:horizontal count "\t"
A [{A}]
C [{C}]
G [{G}]
T [{T}]
%%
Example #4: TRANSFAC format
The header of the TRANSFAC motif format is split across three lines with the use of the newline character "\n". The repeat block used to format the vertically oriented matrix includes both the row number in double digits "{rowXX}" and the consensus letter in upper case "{X}".
AC {ID}
XX
ID {name}
XX
NA {Clean short name}
XX
P0 A C G T
%matrix: vertical
{rowXX} {A} {C} {G} {T} {X}
%%
XX
//
Example #5: XMS format
XMS is an XML-based motif format where normalized frequency matrices are output in a vertical orientation. All the <motif> elements are enclosed in an outer <motifset> tag defined by HEADER and FOOTER.
FOOTER:</motifset>
<motif>
<name>{Short name}</name>
<weightmatrix alphabet="DNA" columns="{size}">
%matrix: vertical frequency
<column pos="{row-1}">
<weight symbol="thymine">{T}</weight>
<weight symbol="guanine">{G}</weight>
<weight symbol="cytosine">{C}</weight>
<weight symbol="adenine">{A}</weight>
</column>
%%
</weightmatrix>
<prop>
<key>Motif_ID</key>
<value>{ID}</value>
</prop>
</motif>
Arguments
| Name | Description |
|---|---|
| Template | The Text Variable containing the output template for formatting a motif, module or sequence. |
| If missing |
This parameter controls how to behave when a motif/module/sequence does not have a defined
value for a particular property referenced in the template. The available options are "Leave blank",
which will just replace the property reference itself with an empty string, or "Skip line" which will
omit the whole line (or even whole repeat block) containing the property reference from the output. For example, consider a template for formatting motifs containing the following three lines: Motif ID = {ID} Class = {Transfac class} Name = {Short name} Now assume that the JASPAR motif "MA0004 - Arnt" does not have a defined value for the "Transfac class" property. If the "If missing" option was set to "Leave blank", the output for this motif would be: Motif ID = MA0004 Class = Name = Arnt However, if the "If missing" option was set to "Skip line", the output would instead be: Motif ID = MA0004 Name = Arnt |
| Sort by | If this optional parameter is specified, the entries in the input collection will be sorted according to their ascending values in this map. If unspecified, the entries will be sorted in natural order. |
See also: output, Motif_Properties, Module_Properties, Sequence_Properties, Template, Text Variable
Location
| Applies to: | Sequence and Sequence Collection |
10-field format
- Sequence identifier
- Organism (taxonomy ID)
- Genome build
- Chromosome
- Start of sequence segment (genomic coordinate)
- End of sequence segment (genomic coordinate)
- Gene name (this could be the same as the sequence ID)
- Transcription start site for the gene (genomic coordinate). This is optional and can be set to NULL
- Transcription end site for the gene (genomic coordinate). This is optional and can be set to NULL
- The strand orientation of the sequence/gene. (DIRECT, +1, +) or (REVERSE, -1, -)
Examples
ENSG00000187664 9606 hg18 19 19244979 19247178 HAPLN4 19245178 19226557 Reverse
ENSG00000196358 9606 hg18 9 134025155 134027354 NTNG2 134027155 134109742 -
8-field format
The 8-field format is a subset of the columns in the 10-field format with the organism identifier and gene name omitted.
The organism is then derived from the genome build and the gene name is set to the same as the sequence identifier.
- Sequence identifier
- Genome build
- Chromosome
- Start of sequence segment (genomic coordinate)
- End of sequence segment (genomic coordinate)
- Transcription start site for the gene (genomic coordinate). This is optional and can be set to NULL
- Transcription end site for the gene (genomic coordinate). This is optional and can be set to NULL
- The strand orientation of the sequence/gene. (DIRECT, +1, +) or (REVERSE, -1, -)
Examples
6-field format
This format specifies the locations of sequences relative known genes. Note that this format can only be used as input.
- Gene identifier (depending on type)
- Name of gene identifier type (e.g. "Ensembl Gene", "Entrez Gene" or "HGNC Symbol")
- Genome build
- Start of sequence segment relative to anchor point (see field #6)
- End of sequence segment relative to anchor point (see field #6)
- Anchor point. This could either be "TSS", "TES". MotifLab v2 also supports the anchor "full gene".
Examples
4-field format
- Sequence identifier
- Chromosome
- Start of sequence segment (genomic coordinate)
- End of sequence segment (genomic coordinate)
Custom format
When the "custom format" is used, the fields to output and how to separate them can be specified manually by describing a string containing special field codes. The following field codes are recognized (case-insensitive):
- Sequence name (or just "name")
- Gene name
- Chromosome
- Start
- End
- Relative start
- Relative end
- TSS
- TES
- Strand
- Genome build (or just "build")
- Organism (or "taxonomy")
- Organism name
- Organism latin name
Example: the custom format "Sequence name,chromosome,start,end" will be similar to the standard 4-field format except that commas are used instead of TABs to separate to fields (to insert TABs use the escape code: \t )
Arguments
| Name | Description |
|---|---|
| Format |
The version of the format to use for output. Valid options are:
|
| Custom Format | If "Custom Format" is selected for the "Format" parameter, the format string must be specified with this parameter as described above for the custom format. |
| add CHR prefix | If this option is selected, a "chr" prefix will be added to the chromosome number when outputting chromosomes. |
| Strand notation | This parameter specifies what notation to use to output the strand orientation of sequences. Valid options are "Direct/Reverse", "+1/-1" or "+/-". |
See also: output
Sequence_Properties
| Applies to: | Sequence and Sequence Collection |
Example of properties output with the format string "name, gene name, organism name, chromosome, genomic start, genomic end" (with header)
Arguments
| Name | Description |
|---|---|
| Format |
This parameter specifies which sequence properties to include in the output
as an ordered, comma-separated list of property names.
Note that standard properties are case-insensitive, but user-defined
properties are case-sensitive! Standard sequence properties include:
|
| Separator | This parameter specifies how to separate the sequence properties on each line in the output. The character chosen as the separator will replace the commas in the format string above. Valid options for the separator are "TAB" (default), "Comma", "Semicolon", "Colon" and "Vertical bar". |
| List-separator | Some sequence properties may be represented by lists (e.g. GO-terms). The "List-separator" parameter specifies how to separate the entries in such lists. Valid options are "Comma" (default), "TAB", "Semicolon", "Colon" and "Vertical bar". Note that this separator should be different from the separator used to separate the sequence properties that are output. |
| Header | If this option is selected, a header line starting with # will be included at the beginning of the output. The header line describes which sequence properties are included. |
See also: output, Motif_Properties, Module_Properties, Properties
Graph
| Applies to: | Text Variable and Output Data |
Examples showing how to dynamically assemble a data table and use this as the basis for creating box plots and histograms: #1 , #2.
Data format requirements
| Chart | Data format |
|---|---|
| Line chart | All formats are accepted. If the input is in "lines" format, only the first two numeric values from each line will be used to define a data point (x,y) and multiple points must be specified on separate lines. |
| Pie chart | All formats are accepted. Only the first value is used from each series. The pie segments are scaled accordingly. |
| Bar chart | All formats are accepted but "lines" is required if bars are to include standard deviation whiskers (since these must be provided as a third value after X (category) and Y. |
| Histogram | All formats are accepted. This chart type ignores dimensionality of data points and will derive the histogram based on all values associated with each series. |
| Box plot | The input data should be in "lines" format with at least 5 values for each series that specify respectively the "minimum" value (bottom whisker), "first quartile" (bottom of box), "median",
"third quartile" (top of box) and "max" value (top whisker). If at least 7 values are provided, the 6th value should be the "average" and the 7th should be the "standard deviation". Any additional values after
these seven will be regarded as outlier values. However, if the "percentiles" plot setting is specified, the statistics will instead be derived dynamamically based on all the values associated with each series. |
Arguments
| Name | Description |
|---|---|
| Graph type |
|
| Data format |
This parameter specifies how the data in the OutputData or TextVariable is formatted.
Three different formats are recognized:
For example, the "line chart" only uses values for the first two dimensions (X,Y) even if more values are provided. For the "histogram" graph type, the dimensionality of the data points have no meaning and the distribution will be based on all values provided for each series. |
| Plot settings |
A semi-colon separated list of key=value pairs that control various aspects of the plot.
The most important settings to specify are the colors to use for drawing each series in the chart, since series that do not have explicitly assigned colors will always be drawn in black.
Colors and style can be defined with
Note that it is possible to draw one or more series as "scatter plots" by setting their linestyle to none and the pointstyle to any other style besides none in line charts. Color and styles for different series can alternatively be specified with display settings on the form: $setting("graph.<property>.<seriesname>")=value (where <property> is either "color", "linestyle" or "pointstyle") Additional plot settings include:
|
| Image width | The original width of the graph image that will be created (before scaling). |
| Image height | The original height of the graph image that will be created (before scaling). |
| Image scale | A scaling factor for the image. The size of the final image will be [width*scale, height*scale]. An image of size [1200,800] at 100% will have the same final size as an image with size [600,400] at 200% scale, but for the second image all the lines will be twice as thick and the text will be twice as big. |
See also: output, Text Variable, Output Data
Plain
| Applies to: | All data objects |
Arguments
| Name | Description |
|---|---|
| Skip comments | This optional parameter can specify a character or text-prefix that denote the beginning of a comment line in the input (e.g. '#' or '//' ). When parsing input in Plain format, all lines that start with the specified prefix will be ignored. |
See also: output, Data