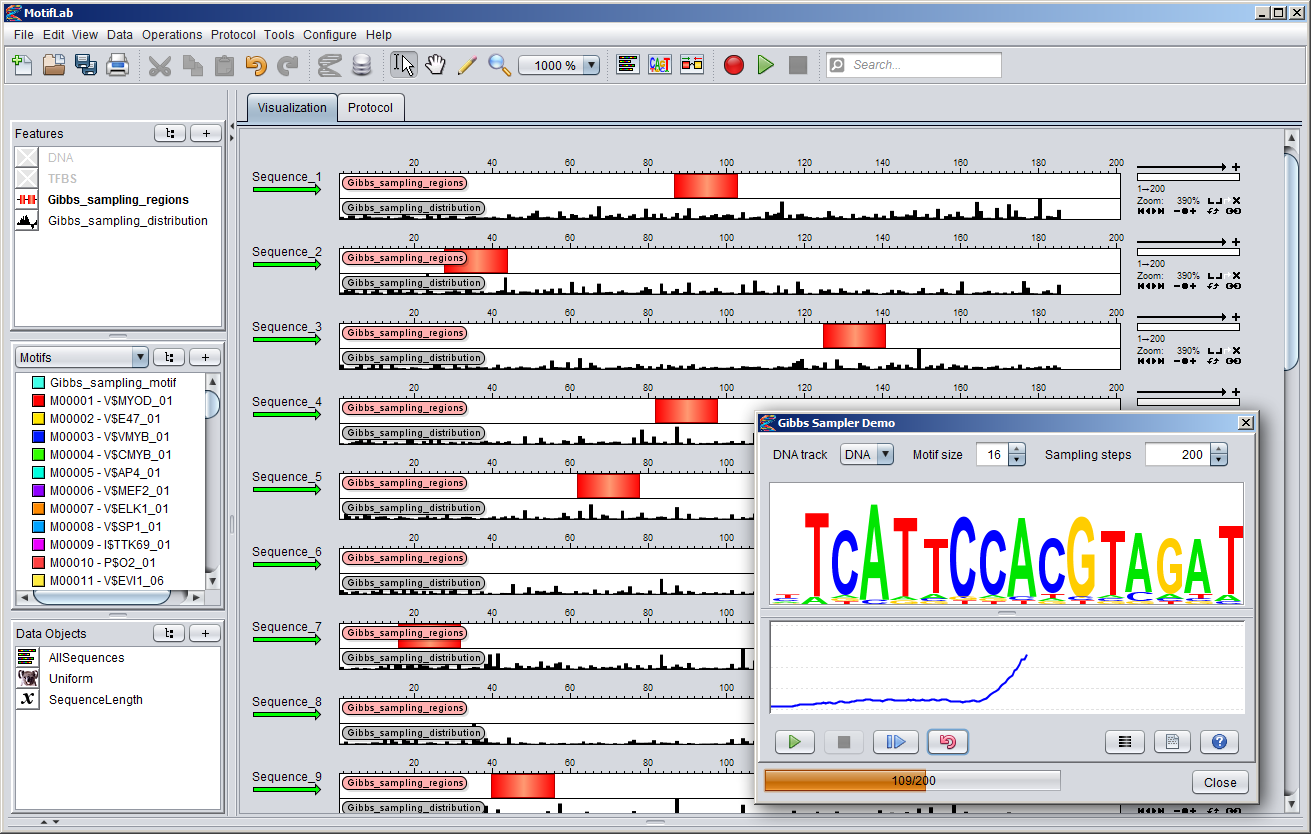
Gibbs Sampler Demo
The "Gibbs Sampler Demo" is an interactive implementation of the popular Gibbs Sampler motif discovery method first proposed by Lawrence et al. [1].
It is a nice educational tool for understanding how motif discovery works in practice since it visualizes every step in the process.
The Gibbs Sampler motif discovery algorithm
The Gibbs Sampler algorithm is an example of an alignment based approach to motif discovery.
In contrast with e.g. word counting based algorithms that exhaustively enumerate every possible DNA sequence of a given length
to find significantly overrepresented motifs in a sequence set, the Gibbs Sampler treats motif discovery as an optimization problem by starting off with an initial model and then
iteratively tries to improve this model using a stochastic hill-climbing approach.
-
The algorithm starts off by selecting a random TFBS site from each sequence and builds a matrix model based on those sites.
This initial model is most likely very bad, since we would not assume that the randomly selected sites share a common motif.
A background model is also created based on the composition of the DNA sequence outside of the selected TFBS sites.
- The algorithm selects one of the sequences and removes its site from the current model (i.e. it creates a new model based on only the TFBS from the other sequences).
The background model is similarly updated.
- The new motif model is now used to score the selected sequence by calculating a motif match score (relative to the background) for each position in the sequence.
Positions that are more similar to the current model will receive higher scores.
- The match scores are turned into a probability distribution and a new candidate TFBS for the sequence is selected at random according to this distribution.
- The newly selected TFBS is incorporated back into the model.
- Steps 2–5 are repeated until the model converges (or a predetermined number of times).
The sequence in step 2 can either be selected at random or all the sequences can be processed systematically in a round-robin fashion.
In the beginning, the sites sampled by the algorithm will most likely not be much similar to each other, and the resulting motif model will have high variability and therefore low information content.
But if, at any point, the algorithm happens to hit upon the correct binding site in a sequence, the model will be slightly skewed towards that binding motif.
This means that when the next sequence is scored, sites in that sequence which are similar to the previously selected sites will receive slightly higher scores and
will therefore have a higher chance of being selected and incorporated into the model during the random sampling step, thus skewing the model even more towards the sites
are similar to the ones already in the model. Hopefully, the algorithm will over time incorporate enough similar sites so that the model eventually converges to the correct motif.
Gibbs Sampler Demo
You can start the "Gibbs Sampler Demo" by selecting it from MotifLab's "Tools" menu. This will bring up the dialog shown below.
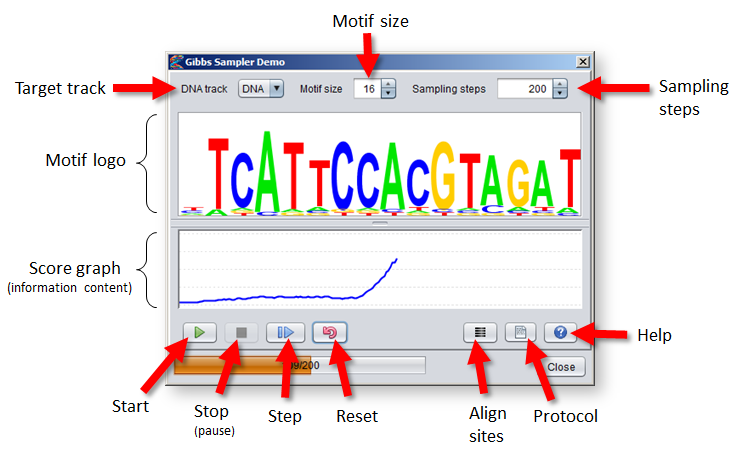
- The DNA track to perform motif discovery in is selected from the menu in the top-left corner of the dialog.
- Select a size for the motif model from the spinner in the middle. The size of the motif can severly influence the performance of the algorithm, so it can be wise to try out different sizes.
Note that the size cannot be adjusted after the Gibbs Sampler has started, so you will have to "reset" it in order to change the size.
- Select the maximum number of sampling steps (iterations) the algorithm will go through before stopping from the spinner to the right.
The method will not stop automatically when the model converges, but will only stop once the maximum number of steps have been reached (or if the stop button is pressed).
- A logo visualization of the current motif model is shown in the middle of the dialog. Right-click on the logo to adjust its settings.
- The score graph beneath the motif logo shows the normalized information content of the motif at each iteration (a value between 0 and 2).
- Pressing the "Start" button will run the algorithm for the given number of sampling steps (or until the "Stop" button is pressed).
At each iteration, the method will select a new sequence, update the motif and background models based on the other sequences,
score the selected sequence using the new motif model and randomly select a new TFBS from the sequence to incorporate back into the motif model.
The currently selected sites in each sequence are stored in a region track called "Gibbs_sampling_regions" and the match scores are stored in a numeric track called "Gibbs_sampling_distribution".
The label of the currently selected sequence is drawn in red color (whereas the others are black). The algorithm is applied to each sequence in a round-robin fashion and will
start over again from the first sequence after all sequences have been processed.
- Pressing the "Stop" button will stop (or rather pause) the algorithm if it has been started. You can press "Start" or "Step" to proceed again from the point where it was stopped.
- Pressing the "Step" button will perform one full iteration of the Gibbs Sampler algorithm (steps 2–5).
- The "Reset" button will discard the current motif model and selected sites to allow the algorithm to be restarted from the beginning.
- Pressing the "Align sites" button will reposition the viewport of each sequence so that the sites in the "Gibbs_sampling_regions" track are aligned in the middle of the sequence windows.
This works best if all sequences have the same zoom level.
- Pressing the "Protocol" button will bring up a protocol script that you can use to create example datasets for motif discovery, as described below.
- The "Help" button brings up a tab showing this documentation page.
Creating an example dataset for motif discovery
The Gibbs Sampler can be applied to search for binding sites in a DNA track for any set of sequences with a common motif.
If you don't already have such a dataset, you can create an example sequence set with the supplied protocol.
Press the "protocol" button in the Gibbs Sampler Demo dialog to bring up the protocol, and then press the "Execute" button in MotifLab's tool bar to run it.
If you have existing sequences you should delete them before you start by selecting "Clear Data" ⇒ "Sequences and Features" from the "Data" menu.
The protocol will first ask you to choose a length for the sequences (the default is 200 bp).
Next, the protocol imports the TRANSFAC PUBLIC motif collection and asks you to choose one of the motifs to plant in the sequences.
Remember to check off the box in front of the motif you want before pressing "OK" in the motif collection dialog, since just selecting the motif name is not enough.
The protocol will then plant the selected motif in the middle of each sequence and return a DNA track and a region track with the target sites (TFBS).
By default, the protocol will create 25 sequences and plant the exact same binding motif in all of the sequences, but it is possible to adjust the total number of sequences,
the number of sequences with planted binding motifs and the variability of the binding motif by editing the protocol.
Tips and tricks
- You can fit more sequences into the Visualization panel by selecting "Show Condensed" from the "View" menu in MotifLab.
- Also, you can change the height of tracks by selecting the tracks in the Features Panel and pressing the +/- buttons (or using the mouse wheel).
If you hold down the CONTROL key at the same time you can adjust the spacing between the sequences.
- If the largest values in the score distributions are very high for some sequences compared to the rest, it can be difficult to see the scores in the lowest scoring sequences.
This can be ameliorated by right-clicking on the "Gibbs_sampling_distribution" track in the Features Panel, selecting "Set Range" and then checking off the "Fit range to each sequence individually" box.
- The SNP Highlighter (available as a plugin) is a perfect tool to emphasize the location of selected regions.
Choose a black color for the highlight, then invert it and adjust the transparency to fade out the background.
Some suggested uses for this tool are:
- Apply the SNP highlighter to the target "TFBS" track but only show the "Gibbs_sampling_regions" track. The location of the target sites will be indicated by a white strip over the track with the currently selected sites as shown in the leftmost image below.
- Apply the SNP highlighter to the "Gibbs_sampling_regions" track but only show the "DNA" track. The currently selected binding sites will stand out brightly against the background DNA as seen in the rightmost image below .

Charles E Lawrence, Stephen F Altschul, Mark S Boguski, Jun S Liu, Andrew F Neuwald and John C Wootton (1993)
"Detecting Subtle Sequence Signals: A Gibbs Sampling Strategy for Multiple Alignment",
Science 262(5131) : 208-214